
Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Penyakit Huntington
Ahli medis artikel

Terakhir ditinjau: 05.07.2025
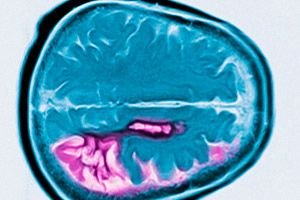
Penyakit Huntington adalah kelainan neurodegeneratif autosomal dominan yang ditandai dengan penurunan kognitif progresif, gerakan tak terkendali, dan gangguan koordinasi motorik yang dimulai pada usia paruh baya. Diagnosis dipastikan dengan pengujian genetik. Pengobatan utamanya bersifat simptomatik. Pengujian genetik mungkin direkomendasikan untuk kerabat sedarah. George Huntington pertama kali mendeskripsikan kondisi tersebut pada tahun 1872, setelah mempelajari kasus keluarga pada penduduk Long Island.
Prevalensi penyakit Huntington adalah sekitar 10 kasus per 100.000 populasi, dan mengingat onsetnya yang lambat, sekitar 30 orang dari 100.000 memiliki risiko 50% untuk mengembangkannya dalam hidup mereka. Meskipun penyakit ini paling sering muncul antara usia 35 dan 40 tahun, rentang usia onsetnya cukup lebar, dengan onset paling awal pada usia 3 tahun dan paling lambat pada usia 90 tahun. Meskipun penyakit ini awalnya dianggap memiliki penetrasi 100%, sekarang diyakini bahwa ini tidak selalu terjadi. Pada individu yang mewarisi gen penyakit dari ayah mereka, penyakit ini memanifestasikan dirinya rata-rata 3 tahun lebih awal daripada pada mereka yang mewarisi gen patologis dari ibu mereka. Pada sekitar 80% pasien yang mewarisi gen patologis dari ayah mereka, penyakit ini memanifestasikan dirinya sebelum usia 20 tahun. Fenomena manifestasi lebih awal dari cacat genetik pada keturunannya disebut antisipasi.
 [ 1 ]
[ 1 ]
Apa penyebab penyakit Huntington?
Penyakit Huntington tidak memiliki preferensi jenis kelamin. Atrofi nukleus kaudatus terlihat, di mana neuron kecil mengalami degenerasi dan kadar neurotransmitter - asam gamma-aminobutyric (GABA) dan substansi P - menurun.
Gen mutan dengan peningkatan jumlah ("ekspansi") urutan DNA CAG (sistein-alanin-glisin) yang mengkode asam amino glutamin bertanggung jawab atas perkembangan penyakit Huntington. Produk gen ini, protein besar huntingtin, mengandung residu poliglutamin dalam jumlah berlebihan, yang menyebabkan penyakit melalui mekanisme yang tidak diketahui. Semakin banyak pengulangan CAG, semakin dini penyakit tersebut muncul dan semakin parah perjalanannya. Dari generasi ke generasi, jumlah pengulangan dapat meningkat, yang seiring waktu menyebabkan perburukan fenotipe keluarga.
Meskipun ada minat yang besar terhadap perubahan genetik dan biokimia pada penyakit Parkinson, pencarian gen untuk penyakit tersebut tidak berhasil hingga akhir tahun 1970-an. Pada saat itu, Nancy Wexler dan Allan Tobin menyelenggarakan lokakarya yang disponsori oleh Hereditary Disease Foundation untuk membahas strategi menemukan gen untuk penyakit Huntington. David Housman, David Botstein, dan Ray White, yang menghadiri pertemuan tersebut, menyarankan bahwa teknik DNA rekombinan yang baru-baru ini dikembangkan dapat membantu mencapai tujuan ini. Tugas utama dalam proyek tersebut adalah menemukan keluarga besar dengan banyak generasi penyakit Huntington untuk mendapatkan sampel DNA. Pada tahun 1979, sebuah proyek gabungan ilmuwan dari Venezuela dan Amerika Serikat diluncurkan untuk memeriksa keluarga besar dengan penyakit Huntington yang tinggal di tepi Danau Maracheibo (Venezuela). Pada tahun 1983, gen penyakit Huntington terlokalisasi di ujung lengan pendek kromosom 4 (Gusella et al., 1983), dan satu dekade kemudian terungkap bahwa mutasi gen ini terdiri dari peningkatan jumlah pengulangan trinukleotida sitosin-adenin-guanin (CAG) (Huntington's Disease Collaborative Research Group, 1993). Metodologi yang dikembangkan oleh kelompok ilmiah ini saat ini dianggap standar untuk kloning posisional gen baru.
Sementara gen tipe liar memiliki rentang 10-28 pengulangan CAG, bentuk mutan dari gen yang menyebabkan penyakit Huntington memiliki rentang yang meningkat dari 39 menjadi lebih dari 100 pengulangan CAG. Penemuan perluasan pengulangan trinukleotida telah membantu menjelaskan banyak fitur klinis penyakit ini. Secara khusus, korelasi terbalik ditemukan antara usia onset dan panjang wilayah dengan trinukleotida berulang. Antisipasi pewarisan paternal dapat dijelaskan oleh fakta bahwa peningkatan jumlah pengulangan sering terjadi pada pria selama spermatogenesis. Analisis mutasi baru telah menunjukkan bahwa mutasi biasanya terjadi ketika salah satu orang tua, biasanya ayah, memiliki jumlah pengulangan CAG lebih tinggi dari 28; dalam kasus ini, jumlah pengulangan ini meningkat pada generasi berikutnya. Sekarang telah ditetapkan bahwa jika jumlah pengulangan tidak lebih dari 28, itu diturunkan secara stabil dari generasi ke generasi. Jika jumlah pengulangannya dari 29 hingga 35, maka gejala penyakit Huntington tidak muncul, tetapi ketika diturunkan ke keturunan, panjang wilayah ini dapat meningkat. Jika jumlah pengulangannya dari 36 hingga 39, maka dalam beberapa kasus (tetapi tidak selalu) penyakit tersebut dapat bermanifestasi secara klinis (penetrasi tidak lengkap), dan ketika diturunkan ke keturunan, peningkatan jumlah pengulangan trinukleotida mungkin terjadi. Jika jumlah pengulangan melebihi 40, maka penyakit tersebut terjadi pada hampir semua kasus, dan ketika diturunkan ke keturunan, perluasan pengulangan lebih lanjut mungkin terjadi. Alasan peningkatan jumlah pengulangan masih belum diketahui.
Patomorfologi penyakit Huntington
Penyakit Huntington ditandai dengan hilangnya neuron terutama di nukleus kaudatus dan putamen, dan sampai batas tertentu juga di korteks dan struktur otak lainnya. Berat total otak pada penyakit Huntington berkurang tidak hanya karena berkurangnya jumlah neuron, tetapi juga karena hilangnya materi putih. Di korteks serebral, sel-sel di lapisan V dan VI paling terpengaruh. Tingkat keparahan perubahan degeneratif mikro dan makroskopis (disesuaikan dengan usia saat kematian) berkorelasi dengan jumlah pengulangan CAG. Analisis patologis terperinci tentang perubahan dalam beberapa ratus kasus penyakit Huntington telah menunjukkan bahwa degenerasi striatum dimulai di bagian dorsomedial nukleus kaudatus dan bagian dorsolateral putamen, dan kemudian menyebar ke bagian ventral. Berbagai kelompok neuron di nukleus kaudatus dan putamen terpengaruh pada tingkat yang berbeda. Interneuron di striatum tetap relatif utuh, tetapi beberapa neuron proyeksi terpengaruh secara selektif. Pada bentuk penyakit Huntington remaja, perubahan patomorfologi di striatum lebih jelas dan lebih luas, melibatkan korteks serebral, serebelum, talamus, dan globus pallidus.
Perubahan neurokimia pada penyakit Huntington
GABA. Studi neurokimia otak pada pasien dengan penyakit Huntington mengungkapkan penurunan yang signifikan dalam konsentrasi GABA di striatum. Studi selanjutnya menegaskan bahwa penyakit Huntington dikaitkan dengan penurunan jumlah neuron GABAergik dan menunjukkan bahwa konsentrasi GABA berkurang tidak hanya di striatum tetapi juga di zona proyeksinya - segmen eksternal dan internal globus pallidus dan substantia nigra. Di otak pada penyakit Huntington, perubahan reseptor GABA juga dideteksi menggunakan studi pengikatan reseptor dan hibridisasi mRNA in situ. Jumlah reseptor GABA berkurang secara moderat di nukleus kaudatus dan putamen, tetapi meningkat di bagian retikuler substantia nigra dan segmen eksternal globus pallidus, yang kemungkinan besar disebabkan oleh hipersensitivitas denervasi.
Asetilkolin. Asetilkolin digunakan sebagai neurotransmitter oleh interneuron nonspiny besar di striatum. Studi postmortem awal pada pasien dengan penyakit Huntington menunjukkan penurunan aktivitas kolin asetiltransferase (ChAT) di striatum, yang menunjukkan hilangnya neuron kolinergik. Namun, dibandingkan dengan penurunan signifikan neuron GABAergik, interneuron kolinergik relatif terhindar. Oleh karena itu, kepadatan neuron asetilkolinesterase-positif dan aktivitas ChAT di striatum sebenarnya relatif meningkat dibandingkan dengan kontrol yang sesuai usia.
Substansi P. Substansi P terkandung dalam banyak neuron spinosus sedang di striatum, yang sebagian besar berproyeksi ke segmen internal globus pallidus dan substansia nigra dan biasanya juga mengandung dinorfin dan GABA. Kadar substansi P di striatum dan pars retikularis substansia nigra berkurang pada penyakit Huntington. Pada tahap terminal penyakit, studi imunohistokimia telah mengungkapkan pengurangan signifikan dalam jumlah neuron yang mengandung substansi P. Pada tahap awal, neuron yang mengandung substansi P dan berproyeksi ke segmen internal globus pallidus relatif lebih sedikit, dibandingkan dengan neuron yang berproyeksi ke pars retikularis substansia nigra.
Peptida opioid. Enkephalin terkandung dalam neuron GABAergik proyeksi spiny sedang dari jalur tidak langsung, yang memproyeksikan ke segmen eksternal globus pallidus dan membawa reseptor D2. Studi imunohistokimia telah menunjukkan bahwa neuron yang mengandung enkephalin yang memproyeksikan ke segmen eksternal globus pallidus hilang lebih awal pada penyakit Huntington. Sel-sel ini tampaknya mati lebih awal daripada sel-sel yang mengandung substansi P yang memproyeksikan ke segmen internal globus pallidus.
Katekolamin. Neuron yang mengandung amina biogenik (dopamin, serotonin) dan berproyeksi ke striatum terletak di bagian kompak substantia nigra, tegmentum ventral, dan nukleus raphe. Sementara proyeksi noradrenergik ke striatum manusia minimal, kadar serotonin dan dopamin (per gram jaringan) di striatum meningkat, yang menunjukkan pelestarian proyeksi aferen ini meskipun neuron striatum sendiri telah hilang secara nyata. Neuron dopaminergik dari substantia nigra tetap utuh dalam bentuk Huntington klasik dan juvenil.
Somatostatin/neuropeptida Y dan sintetase oksida nitrat. Pengukuran kadar somatostatin dan neuropeptida Y di striatum pada penyakit Huntington menunjukkan peningkatan 4-5 kali lipat dibandingkan dengan jaringan normal. Studi imunohistokimia menunjukkan pelestarian mutlak interneuron striatal yang mengandung neuropeptida Y, somatostatin, dan sintetase oksida nitrat. Dengan demikian, neuron-neuron ini resistan terhadap proses patologis.
Asam amino eksitatori. Telah dikemukakan bahwa kematian sel selektif pada penyakit Huntington disebabkan oleh efek neurotoksik yang disebabkan oleh glutamat. Kadar glutamat dan asam quinolinic (neurotoksin endogen yang merupakan produk sampingan metabolisme serotonin dan agonis reseptor glutamat) di striatum penyakit Huntington sedikit berubah, tetapi penelitian terbaru menggunakan spektroskopi MR mengungkapkan peningkatan kadar glutamat in vivo. Kadar enzim glia yang bertanggung jawab untuk sintesis asam quinolinic di striatum penyakit Huntington meningkat sekitar 5 kali lipat dibandingkan dengan normal, sementara aktivitas enzim yang memastikan degradasi asam quinolinic meningkat pada penyakit Huntington hanya sebesar 20-50%. Dengan demikian, sintesis asam quinolinic dapat meningkat pada penyakit Huntington.
Studi reseptor asam amino eksitatori (EAA) pada penyakit Huntington telah mengungkap pengurangan signifikan jumlah reseptor NMDA, AMPA, kainat, dan glutamat metabotropik di striatum, serta reseptor AMPA dan kainat di korteks serebral. Pada tahap akhir penyakit Huntington, reseptor NMDA hampir tidak ada, sedangkan pada tahap praklinis dan awal, pengurangan signifikan jumlah reseptor ini dicatat.
Sensitivitas selektif. Pada penyakit Huntington, beberapa jenis sel striatal hilang secara selektif. Neuron spinosus sedang, yang berproyeksi ke segmen luar globus pallidus dan mengandung GABA dan enkephalin, mati sangat dini pada penyakit ini, seperti halnya neuron yang mengandung GABA dan substansi P dan berproyeksi ke bagian retikuler substantia nigra. Hilangnya neuron yang mengandung GABA dan enkephalin dan berproyeksi ke segmen luar globus pallidus menghilangkan hambatan pada struktur ini, yang pada gilirannya menyebabkan penghambatan aktif nukleus subthalamik. Penurunan aktivitas nukleus subthalamik tampaknya dapat menjelaskan gerakan koreiform yang terjadi pada penyakit Huntington. Telah lama diketahui bahwa lesi fokal pada nukleus subthalamik dapat menyebabkan koreo. Hilangnya neuron GABA dan substansi P yang berproyeksi ke substantia nigra pars reticularis kemungkinan besar bertanggung jawab atas gangguan okulomotor yang terlihat pada penyakit Huntington. Jalur ini biasanya menghambat neuron substantia nigra pars reticularis yang memproyeksikan ke kolikulus superior, yang selanjutnya mengatur gerakan mata. Pada penyakit Huntington juvenil, jalur yang disebutkan di atas lebih parah terpengaruh dan, sebagai tambahan, proyeksi striatal ke segmen internal globus pallidus hilang lebih awal.
Protein huntingtin, yang dikodekan oleh gen yang mutasinya menyebabkan penyakit Huntington, ditemukan dalam berbagai struktur otak dan jaringan lainnya. Huntingtin biasanya ditemukan terutama dalam sitoplasma neuron. Protein ini ditemukan di sebagian besar neuron di otak, tetapi data terkini menunjukkan bahwa kandungannya lebih tinggi dalam neuron matriks daripada dalam neuron striosomal, dan lebih tinggi dalam neuron proyeksi daripada dalam interneuron. Dengan demikian, sensitivitas selektif neuron berkorelasi dengan kandungan huntingtinnya, yang biasanya terdapat dalam populasi neuron tertentu.
Seperti pada otak pasien dengan penyakit Huntington, pada tikus yang ditransgenik untuk fragmen N-terminal gen penyakit Huntington dengan jumlah pengulangan yang lebih banyak, huntingtin membentuk agregat padat di dalam nukleus neuron. Inklusi intranukleus ini terbentuk di neuron proyeksi striatal (tetapi tidak di interneuron). Pada tikus transgenik, inklusi terbentuk beberapa minggu sebelum timbulnya gejala. Data ini menunjukkan bahwa protein huntingtin yang mengandung lebih banyak residu glutamin yang inklusinya mengkode pengulangan trinukleotida, atau sebagian darinya, terakumulasi di dalam nukleus dan akibatnya dapat mengganggu kontrolnya terhadap fungsi seluler.
Gejala Penyakit Huntington
Usia saat gejala pertama kali muncul pada pasien dengan penyakit Huntington sulit ditentukan dengan tepat, karena penyakit ini bermanifestasi secara bertahap. Perubahan kepribadian dan perilaku, gangguan koordinasi ringan dapat terjadi bertahun-tahun sebelum munculnya gejala yang lebih jelas. Pada saat diagnosis ditegakkan, sebagian besar pasien mengalami gerakan koreik, gangguan koordinasi gerakan halus, dan lambatnya pembentukan gerakan sadar. Seiring perkembangan penyakit, kemampuan mengatur aktivitas terganggu, daya ingat menurun, bicara menjadi sulit, gangguan okulomotor dan gangguan kinerja gerakan terkoordinasi meningkat. Meskipun pada tahap awal penyakit tidak ada perubahan pada otot dan postur, seiring perkembangannya, postur distonik dapat berkembang, yang seiring waktu dapat berubah menjadi gejala yang dominan. Pada tahap akhir, bicara menjadi tidak jelas, menelan menjadi sangat sulit, berjalan menjadi tidak mungkin. Penyakit Huntington biasanya berkembang selama 15-20 tahun. Pada tahap terminal, pasien tidak berdaya dan membutuhkan perawatan terus-menerus. Hasil yang fatal tidak terkait langsung dengan penyakit primer, tetapi dengan komplikasinya, misalnya, pneumonia.
Demensia pada penyakit Huntington
Kode ICD-10
P02.2. Demensia pada penyakit Huntington (G10).
Demensia berkembang sebagai salah satu manifestasi dari proses degeneratif-atrofi sistemik dengan kerusakan dominan pada sistem striatal otak dan nukleus subkoekal lainnya. Penyakit ini diwariskan secara autosomal dominan.
Penyakit ini biasanya memanifestasikan dirinya pada dekade ketiga atau keempat kehidupan dengan hiperkinesis koreoform (terutama pada wajah, lengan, bahu, gaya berjalan), perubahan kepribadian (anomali kepribadian tipe bersemangat, histeris, dan skizoid), gangguan psikotik (depresi khusus dengan kesuraman, cemberut, disforia; suasana hati paranoid).
Yang paling penting untuk diagnostik adalah kombinasi hiperkinesis koreoform, demensia, dan beban keturunan. Berikut ini adalah hal-hal khusus untuk demensia ini:
- perkembangan yang lambat (rata-rata 10-15 tahun): disosiasi antara kemampuan yang tersisa untuk mengurus diri sendiri dan inkompetensi intelektual yang jelas dalam situasi yang membutuhkan kerja mental yang produktif (berpikir konseptual, mempelajari hal-hal baru);
- ketidakseimbangan kinerja mental yang nyata, yang didasarkan pada gangguan perhatian yang parah dan ketidakkonsistenan sikap pasien (pemikiran "sentak", mirip dengan hiperkinesis);
- atipikalitas pelanggaran yang jelas terhadap fungsi kortikal yang lebih tinggi;
- hubungan terbalik antara peningkatan demensia dan tingkat keparahan gangguan psikotik.
Mengingat tingginya proporsi gangguan psikotik (delusi paranoid seperti cemburu, penganiayaan) dan gangguan disforik pada gambaran klinis penyakit ini, pengobatan dilakukan dengan menggunakan berbagai neuroleptik yang memblokir reseptor dopaminergik (turunan fenotiazin dan butirofenon) atau mengurangi kadar dopamin dalam jaringan (reserpin).
Haloperidol (2-20 mg/hari), tiapride (100-600 mg/hari) selama tidak lebih dari tiga bulan, thioridazine (hingga 100 mg/hari), reserpine (0,25-2 mg/hari), dan antikonvulsan clonazepam (1,5-6 mg/hari) digunakan. Obat-obatan ini membantu mengurangi hiperkinesis, meredakan ketegangan afektif, dan mengimbangi gangguan kepribadian.
Perawatan gangguan mental di rumah sakit dilakukan dengan mempertimbangkan sindrom yang mendasarinya, usia, dan kondisi umum pasien. Dalam perawatan rawat jalan, prinsip terapinya sama (terapi pemeliharaan berkelanjutan untuk gangguan gerakan, penggantian obat secara berkala). Dosis neuroleptik yang lebih rendah digunakan dalam perawatan rawat jalan.
Tindakan rehabilitasi untuk demensia ringan dan sedang meliputi terapi okupasi, psikoterapi, dan pelatihan kognitif. Perlu bekerja sama dengan anggota keluarga dan memberikan dukungan psikologis kepada orang yang merawat pasien. Metode utama pencegahan penyakit adalah konseling medis dan genetik dari kerabat terdekat pasien dengan rujukan untuk analisis DNA saat memutuskan untuk memiliki anak.
Prognosisnya secara umum tidak baik. Perjalanan penyakitnya lambat dan progresif, dan penyakit ini biasanya menyebabkan kematian setelah 10-15 tahun.
[ 18 ]
Apa yang mengganggumu?
Pengobatan penyakit Huntington
Pengobatan penyakit Huntington bersifat simtomatik. Korea dan agitasi dapat ditekan sebagian dengan neuroleptik (misalnya, klorpromazin 25-300 mg per oral 3 kali sehari, haloperidol 5-45 mg per oral 2 kali sehari) atau reserpin 0,1 mg per oral sekali sehari. Dosis ditingkatkan hingga batas maksimum yang dapat ditoleransi (sebelum efek samping terjadi, seperti kantuk, parkinsonisme; untuk reserpin, hipotensi). Tujuan terapi empiris adalah untuk mengurangi transmisi glutamatergik melalui reseptor N-metil-O-aspartat dan mempertahankan produksi energi dalam mitokondria. Pengobatan yang ditujukan untuk meningkatkan GABA di otak tidak efektif.
Pengujian dan konseling genetik penting karena gejala penyakit muncul setelah masa subur. Orang dengan riwayat keluarga positif dan mereka yang tertarik untuk menjalani pengujian dirujuk ke pusat-pusat khusus, dengan mempertimbangkan semua implikasi etis dan psikologis.
Pengobatan simtomatik penyakit Huntington
Tidak ada pengobatan efektif yang dapat menghentikan perkembangan penyakit Huntington. Beberapa uji coba berbagai obat telah dilakukan, tetapi belum ada efek signifikan yang dicapai. Neuroleptik dan antagonis reseptor dopamin lainnya banyak digunakan untuk mengoreksi gangguan mental dan gerakan tak sadar pada pasien dengan penyakit Huntington. Gerakan tak sadar mencerminkan ketidakseimbangan antara sistem dopaminergik dan GABAergik. Oleh karena itu, neuroleptik digunakan untuk mengurangi aktivitas dopaminergik berlebih. Namun, obat-obatan ini sendiri dapat menyebabkan efek samping kognitif dan ekstrapiramidal yang signifikan. Selain itu, kecuali dalam kasus di mana pasien mengalami psikosis atau agitasi, efektivitasnya belum terbukti. Neuroleptik sering menyebabkan atau memperburuk disfagia atau gangguan gerakan lainnya. Neuroleptik generasi baru seperti risperidone, clozapine, dan olanzapine mungkin sangat berguna dalam pengobatan penyakit Huntington karena obat-obatan ini menyebabkan lebih sedikit efek samping ekstrapiramidal tetapi dapat mengurangi gejala paranoid atau peningkatan iritabilitas.
Tetrabenazin dan reserpin juga mengurangi aktivitas sistem dopaminergik dan dapat mengurangi keparahan gerakan tak sadar pada tahap awal penyakit. Namun, obat-obatan ini dapat menyebabkan depresi. Karena penyakit itu sendiri sering menyebabkan depresi, efek samping ini secara signifikan membatasi penggunaan reserpin dan tetrabenazin. Pada tahap akhir penyakit, sel-sel yang mengandung reseptor dopamin mati, sehingga efektivitas antagonis reseptor dopamin melemah atau hilang.
Neuroleptik, antidepresan, dan ansiolitik digunakan untuk mengobati psikosis, depresi, dan mudah tersinggung pada pasien dengan penyakit Huntington, tetapi obat-obatan tersebut hanya boleh diresepkan selama pasien masih mengalami gejala-gejala tersebut. Obat-obatan yang mungkin bermanfaat pada satu tahap penyakit dapat menjadi tidak efektif atau bahkan berbahaya seiring perkembangan penyakit.
Agonis reseptor GABA telah diuji pada pasien dengan penyakit Huntington, karena penyakit Huntington telah terbukti memiliki penurunan kadar GABA yang signifikan di striatum, serta hipersensitivitas reseptor GABA di area proyeksinya. Benzodiazepin telah terbukti efektif dalam kasus-kasus di mana gerakan tak sadar dan gangguan kognitif diperburuk oleh stres dan kecemasan. Dosis rendah obat-obatan ini harus diresepkan untuk menghindari efek sedatif yang tidak diinginkan. Pada sebagian besar pasien dengan penyakit Huntington, tidak ada obat yang mengarah pada peningkatan kualitas hidup yang signifikan.
Pada penyakit Huntington yang muncul lebih awal dengan gejala parkinson, agen dopaminergik dapat dicoba, tetapi efektivitasnya terbatas. Selain itu, levodopa dapat menyebabkan atau meningkatkan mioklonus pada pasien ini. Pada saat yang sama, baklofen dapat mengurangi kekakuan pada beberapa pasien dengan penyakit Huntington.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Pengobatan pencegahan (neuroprotektif) penyakit Huntington
Meskipun cacat genetik pada penyakit Huntington diketahui, bagaimana hal itu menyebabkan degenerasi neuronal selektif masih belum jelas. Dihipotesiskan bahwa terapi pencegahan yang ditujukan untuk mengurangi stres oksidatif dan eksitotoksisitas berpotensi memperlambat atau menghentikan perkembangan penyakit. Situasinya mungkin agak mirip dengan degenerasi hepatolentikular, di mana cacat genetik tetap tidak diketahui selama bertahun-tahun, tetapi terapi pencegahan yang ditujukan pada efek sekunder, akumulasi tembaga, "disembuhkan." Dalam hal ini, hipotesis bahwa penyakit Huntington dikaitkan dengan gangguan metabolisme energi dan kematian sel akibat eksitotoksisitas telah menarik perhatian khusus. Penyakit itu sendiri dapat menyebabkan kematian sel akibat agregasi intranuklear fragmen N-terminal huntingtin, yang mengganggu fungsi seluler dan metabolisme. Proses ini dapat memengaruhi beberapa kelompok neuron lebih besar daripada yang lain karena sensitivitasnya yang lebih tinggi terhadap kerusakan eksitotoksik. Dalam kasus ini, terapi pencegahan dengan antagonis reseptor asam amino eksitatori atau agen yang mencegah kerusakan radikal bebas akan dapat mencegah atau menunda timbulnya dan perkembangan penyakit. Dalam model laboratorium sklerosis lateral amiotrofik, telah ditunjukkan bahwa agen antioksidan dan antagonis reseptor (RAA) mampu memperlambat perkembangan penyakit. Pendekatan serupa mungkin efektif dalam penyakit Huntington. Uji klinis antagonis reseptor glutamat dan agen yang meningkatkan fungsi kompleks II rantai transpor elektron mitokondria saat ini sedang berlangsung.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]