
Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Sindrom Martin-Bell
Ahli medis artikel

Terakhir ditinjau: 04.07.2025
Sindrom Martin-Bell dideskripsikan pada tahun 1943 oleh para dokter, yang kemudian menamainya demikian. Penyakit ini merupakan kelainan genetik yang terdiri dari keterbelakangan mental. Pada tahun 1969, perubahan pada kromosom X (kerapuhan pada lengan distal) yang menjadi ciri khas penyakit ini diidentifikasi. Pada tahun 1991, para ilmuwan menemukan gen yang bertanggung jawab atas perkembangan penyakit ini. Penyakit ini juga disebut "sindrom fragile X". Baik anak laki-laki maupun perempuan rentan terhadap penyakit ini, tetapi anak laki-laki lebih sering (3 kali) terkena.
Epidemiologi
Sindrom Martin-Bell merupakan penyakit yang cukup umum: 0,3-1,0 dari 1.000 pria menderita penyakit ini, dan 0,2-0,6 dari 1.000 wanita. Selain itu, anak-anak dengan sindrom Martin-Bell lahir di semua benua dengan frekuensi yang sama. Jelas, kebangsaan, warna kulit, bentuk mata, kondisi kehidupan, dan kesejahteraan masyarakat tidak memengaruhi terjadinya penyakit ini. Frekuensi kejadiannya hanya sebanding dengan frekuensi sindrom Down (1 penyakit dari 600-800 bayi baru lahir). Seperlima dari pembawa gen yang diubah laki-laki sehat, tidak memiliki kelainan klinis atau gen, sisanya memiliki tanda-tanda keterbelakangan mental dari bentuk ringan hingga berat. Di antara pembawa perempuan, sedikit lebih dari sepertiganya sakit.
Sindrom Fragile X menyerang sekitar 1 dari 2.500–4.000 pria dan 1 dari 7.000–8.000 wanita. Prevalensi pembawa di antara wanita diperkirakan 1 dari 130–250; prevalensi pembawa di antara pria diperkirakan 1 dari 250–800.
Penyebab Sindrom Martin-Bell
Sindrom Martin-Bell berkembang karena penghentian total atau sebagian produksi protein tertentu oleh tubuh. Hal ini terjadi karena kurangnya respons dari gen FMR1, yang terlokalisasi dalam kromosom X. Mutasi terjadi sebagai akibat dari restrukturisasi gen dari varian struktural yang tidak stabil dari keadaan gen (alel), dan bukan dari awal. Penyakit ini ditularkan hanya melalui garis laki-laki, dan laki-laki tersebut belum tentu sakit. Pembawa laki-laki mewariskan gen kepada anak perempuan mereka dalam bentuk yang tidak berubah, sehingga keterbelakangan mental mereka tidak terlihat. Dengan transmisi gen lebih lanjut dari ibu ke anak-anaknya, gen tersebut bermutasi, dan semua tanda karakteristik penyakit ini muncul.
Patogenesis
Patogenesis sindrom Martin-Bell didasarkan pada mutasi pada aparatus gen, yang menyebabkan terhambatnya produksi protein FMR, protein yang vital bagi tubuh, terutama pada neuron, dan terdapat di berbagai jaringan. Penelitian menunjukkan bahwa protein FMR terlibat langsung dalam proses pengaturan translasi yang terjadi di jaringan otak. Tidak adanya protein ini atau terbatasnya produksinya oleh tubuh menyebabkan keterbelakangan mental.
Dalam patogenesis penyakit ini, hipermetilasi gen dianggap sebagai kelainan utama, tetapi belum dapat diketahui secara pasti mekanisme perkembangan kelainan ini.
Pada saat yang sama, heterogenitas lokus patologi juga ditemukan, yang dikaitkan dengan polialelisme, serta polilokus. Keberadaan varian alel dari perkembangan penyakit ditentukan, yang disebabkan oleh keberadaan mutasi titik, serta kerusakan gen tipe FMRL.
Pasien juga memiliki 2 triplet rapuh yang sensitif terhadap asam folat, terletak 300 kb, serta 1,5-2 juta bp dari triplet rapuh yang mengandung gen FMR1. Mekanisme mutasi yang terjadi pada gen FRAXE dan FRAXF (keduanya diidentifikasi dalam triplet rapuh yang disebutkan di atas) terkait dengan mekanisme kelainan pada sindrom Martin Bell. Mekanisme ini disebabkan oleh penyebaran pengulangan GCC dan CGG, yang menyebabkan metilasi dari apa yang disebut pulau CpG. Selain bentuk patologi klasik, ada juga 2 jenis langka yang berbeda karena perluasan pengulangan trinukleotida (pada meiosis pria dan wanita).
Ditemukan bahwa pada bentuk klasik sindrom ini, pasien kekurangan protein nukleositoplasma khusus jenis FMR1, yang berfungsi mengikat berbagai mRNA. Selain itu, protein ini mendorong pembentukan kompleks yang membantu melaksanakan proses translasi di dalam ribosom.
Gejala Sindrom Martin-Bell
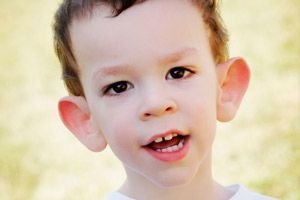
Bagaimana mengenali penyakit ini pada anak-anak? Apa saja tanda-tanda pertamanya? Pada bulan-bulan pertama kehidupan seorang anak, tidak mungkin untuk mengenali gejala Martin-Bell, kecuali bahwa kadang-kadang penurunan tonus otot diamati. Setelah satu tahun, gambaran klinis penyakit ini lebih jelas: anak mulai berjalan dan berbicara terlambat, kadang-kadang bicaranya sama sekali tidak ada. Dia hiperaktif, melambaikan tangannya secara acak, takut pada keramaian dan kebisingan, keras kepala, ada ledakan amarah yang tajam, ketidakstabilan emosi, terjadi kejang epilepsi, tidak melakukan kontak mata. Pada pasien dengan sindrom Martin-Bell, penyakit ini juga terlihat dari penampilannya: telinga menonjol dan besar, dahi berat, wajah memanjang, dagu menonjol, strabismus, tangan dan kaki lebar. Mereka juga ditandai oleh gangguan endokrin: seringkali berat badan besar, obesitas, testis besar pada pria, pubertas dini.

Di antara pasien dengan sindrom Martin-Bell, tingkat kecerdasan sangat bervariasi: dari keterbelakangan mental ringan hingga kasus yang parah. Jika orang normal memiliki kecerdasan intelektual (IQ) rata-rata 100, dan seorang jenius memiliki 130, maka orang yang rentan terhadap penyakit ini memiliki 35-70.
Semua gejala klinis patologi dapat ditandai dengan tiga serangkai manifestasi utama:
- oligofrenia (IQ 35-50);
- dismorfofobia (telinga menonjol dan prognatisme diamati);
- makroorkidisme, yang muncul setelah masa pubertas.
Sekitar 80% pasien juga mengalami prolaps katup bikuspid.
Namun, bentuk lengkap sindrom ini hanya muncul pada 60% dari semua pasien. Pada 10%, hanya retardasi mental yang terdeteksi, dan pada sisanya, penyakit ini berkembang dengan kombinasi gejala yang berbeda.
Di antara tanda-tanda pertama penyakit ini, yang muncul pada usia dini:
- anak yang sakit menunjukkan keterbelakangan mental yang signifikan dibandingkan dengan perkembangan teman sebayanya;
- gangguan perhatian dan konsentrasi;
- keras kepala yang kuat;
- anak-anak mulai berjalan dan berbicara agak terlambat;
- hiperaktivitas dan gangguan perkembangan bicara diamati;
- ledakan kemarahan yang sangat kuat dan tak terkendali;
- mutisme dapat berkembang - ini adalah tidak adanya kemampuan bicara sama sekali pada seorang anak;
- bayi mengalami kecemasan sosial dan mampu panik karena suara keras atau bunyi keras lainnya;
- anak itu melambaikan tangannya tak terkendali dan kacau;
- rasa malu diamati, anak takut berada di tempat ramai;
- munculnya berbagai ide obsesif, keadaan emosi yang tidak stabil;
- Bayi mungkin enggan melakukan kontak mata dengan orang lain.
Pada orang dewasa, gejala patologi berikut diamati:
- penampilan khusus: wajah memanjang dengan dahi tebal, telinga besar menonjol, dagu sangat menonjol;
- kaki datar, otitis dan strabismus;
- pubertas terjadi cukup awal;
- obesitas mungkin berkembang;
- Cukup sering, cacat jantung diamati pada sindrom Martin-Bell;
- pada pria, terjadi pembesaran testis;
- artikulasi sendi menjadi sangat mobile;
- berat badan dan tinggi badan meningkat tajam.
Diagnostik Sindrom Martin-Bell
Untuk mendiagnosis sindrom Martin Bell, Anda perlu menghubungi ahli genetika yang berkualifikasi. Diagnosis dilakukan setelah menjalani tes genetika khusus yang memungkinkan Anda mengidentifikasi kromosom yang rusak.
Tes
Pada tahap awal penyakit, metode sitogenetik digunakan, di mana fragmen materi seluler diambil dari pasien, yang kemudian ditambahkan asam folat untuk memicu perubahan pada kromosom. Setelah jangka waktu tertentu, area kromosom teridentifikasi di mana terdapat penipisan yang nyata - ini merupakan tanda adanya sindrom fragile X.
Akan tetapi, tes ini tidak cocok untuk diagnosis pada stadium lanjut penyakit, karena akurasinya berkurang akibat meluasnya penggunaan multivitamin yang mengandung asam folat.
Diagnostik terpadu sindrom Martin-Bell adalah pemeriksaan genetik molekuler, yang terdiri dari penentuan jumlah yang disebut pengulangan trinukleotida dalam gen.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnostik instrumental
Metode diagnostik instrumental yang sangat spesifik adalah PCR (polymerase chain reaction), yang memungkinkan seseorang mempelajari struktur residu asam amino yang terkandung dalam kromosom X dan dengan demikian menentukan keberadaan sindrom Martin Bell.
Ada pula metode diagnostik patologi yang terpisah dan lebih spesifik, yaitu kombinasi PCR dan deteksi menggunakan elektroforesis kapiler. Metode ini sangat akurat dan mendeteksi patologi kromosom pada pasien dengan kegagalan ovarium primer, serta sindrom ataksik.
Keberadaan kelainan ini dapat diketahui setelah dilakukan diagnostik pada EEG. Pasien dengan penyakit ini memiliki aktivitas bioelektrik otak yang serupa.
Tes apa yang dibutuhkan?
Perbedaan diagnosa
Metode-metode yang dibedakan yang membantu untuk mencurigai adanya sindrom ini meliputi:
- klinis - 97,5% pasien memiliki tanda-tanda retardasi mental yang nyata (sedang atau berat); 62% memiliki telinga yang menonjol dan besar; 68,4% memiliki dagu dan dahi yang menonjol dan besar; 68,4% anak laki-laki memiliki testis yang membesar, 41,4% memiliki kekhasan bicara (kecepatan bicara tidak merata, volume yang tidak terkendali, dll.);
- sitogenik - darah diperiksa untuk kultur limfosit, jumlah sel dengan kromosom X rapuh per 100 sel yang diteliti ditentukan;
- Elektroensefalografi - perubahan impuls listrik otak yang khusus untuk sindrom Martin-Bell direkam.
Siapa yang harus dihubungi?
Pengobatan Sindrom Martin-Bell
Dalam pengobatan pasien dewasa, antidepresan dengan psikostimulan digunakan. Proses terapi obat dipantau terus-menerus oleh psikolog dan psikiater. Selain itu, prosedur mikroinjeksi dengan obat-obatan seperti Cerebrolysin (atau turunannya), serta sitomedin (seperti Solcoseryl atau Lidase) dilakukan di klinik swasta.
Dalam perkembangan sindrom ataksik, obat pengencer darah dan nootropik digunakan. Selain itu, campuran asam amino dan angioprotektor diresepkan. Wanita dengan kegagalan ovarium primer diresepkan pengobatan korektif menggunakan obat-obatan herbal dan estrogen.
Antagonis reseptor glutamin juga digunakan dalam pengobatan.
Secara tradisional, pengobatan sindrom Martin-Bell melibatkan penggunaan obat-obatan yang memengaruhi gejala penyakit, tetapi bukan penyebabnya. Terapi ini melibatkan pemberian resep antidepresan, neuroleptik, dan psikostimulan. Tidak semua obat diindikasikan untuk digunakan pada anak-anak, sehingga daftar obatnya cukup terbatas. Neuroleptik yang dapat digunakan setelah 3 tahun (usia paling awal untuk resepnya) meliputi haloperidol dalam bentuk tetes dan tablet, klorpromazin dalam bentuk larutan, dan periciazine dalam bentuk tetes. Dengan demikian, dosis haloperidol untuk anak-anak dihitung tergantung pada berat badan. Untuk orang dewasa, dosisnya ditentukan secara individual. Obat ini diminum secara oral, dimulai dengan 0,5–5 mg 2–3 kali sehari, kemudian dosisnya ditingkatkan secara bertahap menjadi 10–15 mg. Ketika terjadi perbaikan, dosisnya beralih ke dosis yang lebih rendah untuk mempertahankan kondisi yang dicapai. Dalam kasus agitasi psikomotorik, 5–10 mg diresepkan secara intramuskular atau intravena, beberapa kali pengulangan dapat dilakukan setelah 30–40 menit. Dosis harian tidak boleh melebihi 100 mg. Efek samping berupa mual, muntah, kejang otot, peningkatan tekanan, aritmia, dll. mungkin terjadi. Orang lanjut usia harus mengambil tindakan pencegahan khusus, karena kasus serangan jantung mendadak telah tercatat, dan tardive dyskinesia (gerakan tak sadar) dapat terjadi.
Antidepresan meningkatkan aktivitas struktur otak, meredakan depresi, ketegangan, dan memperbaiki suasana hati. Obat-obatan ini, yang direkomendasikan untuk digunakan pada usia 5-8 tahun untuk sindrom Martin-Bell, meliputi klomipromin, sertralin, fluoksetin, dan fluvoksamin. Jadi, fluoksetin diminum secara oral selama makan 1-2 kali (sebaiknya pada paruh pertama hari), dimulai dengan 20 mg per hari, ditingkatkan menjadi 80 mg jika perlu. Orang lanjut usia tidak direkomendasikan dosis lebih tinggi dari 60 mg. Jalannya pengobatan ditentukan oleh dokter, tetapi tidak lebih dari 5 minggu.
Kemungkinan efek samping: pusing, cemas, tinitus, kehilangan nafsu makan, takikardia, edema, dll. Kehati-hatian diperlukan saat meresepkannya kepada orang tua, mereka yang memiliki penyakit kardiovaskular, dan diabetes.
Psikostimulan adalah obat psikotropika yang digunakan untuk meningkatkan persepsi rangsangan eksternal: mereka mempertajam pendengaran, reaksi respons, dan penglihatan.
Diazepam diresepkan sebagai obat penenang untuk neurosis, kecemasan, kejang epilepsi, dan konvulsi. Obat ini diminum secara oral, intravena, intramuskular, rektal (ke dalam rektum). Obat ini diresepkan secara individual, tergantung pada tingkat keparahan penyakit, dengan dosis terkecil 5-10 mg, setiap hari - 5-20 mg. Durasi pengobatan adalah 2-3 bulan. Untuk anak-anak, dosis dihitung dengan mempertimbangkan berat badan dan karakteristik individu. Efek sampingnya meliputi kelesuan, apatis, kantuk, mual, sembelit. Obat ini berbahaya jika dikombinasikan dengan alkohol, dapat menyebabkan kecanduan.
Dalam pengobatan sindrom Martin-Bell, ada beberapa kasus perbaikan kondisi dengan pemberian obat yang terbuat dari bahan hewani (otak): serebrolisin, serebrolisin, serebrolisin-M. Komponen utama obat ini adalah peptida yang meningkatkan produksi protein dalam neuron, sehingga mengisi kembali protein yang hilang. Serebrolisin diberikan dalam bentuk jet sebanyak 5-10 ml, pengobatannya terdiri dari 20-30 suntikan. Obat ini diresepkan untuk anak-anak sejak usia satu tahun, diberikan secara intramuskular setiap hari sebanyak 1-2 ml selama sebulan. Sesi pemberian berulang dimungkinkan. Efek samping berupa demam, kontraindikasi untuk wanita hamil.
Ada upaya untuk mengobati penyakit dengan asam folat, tetapi hanya aspek perilaku yang membaik (tingkat agresi dan hiperaktif menurun, kemampuan bicara membaik), dan tidak ada yang berubah pada tingkat intelektual. Untuk memperbaiki kondisi penyakit, asam folat diresepkan, metode fisioterapi, terapi wicara, koreksi pedagogis dan sosial diindikasikan.
Sediaan litium juga dianggap efektif, karena membantu meningkatkan adaptasi pasien terhadap lingkungan sosial, serta aktivitas kognitif. Selain itu, sediaan ini juga mengatur perilakunya di masyarakat.
Penggunaan herba untuk sindrom Martin-Bell dapat dilakukan sebagai antidepresan. Herba yang membantu meredakan ketegangan, kecemasan, dan meningkatkan kualitas tidur meliputi valerian, pepermin, timi, St. John's wort, dan kamomil. Infus disiapkan sebagai berikut: untuk 1 sendok teh herba kering, Anda memerlukan segelas air mendidih, rebusan diinfuskan selama setidaknya 20 menit, diminum terutama pada malam hari sebelum tidur atau di sore hari. Sesendok madu akan menjadi tambahan yang baik untuknya.
Perawatan fisioterapi
Untuk menghilangkan manifestasi neurologis, prosedur fisioterapi khusus dilakukan - seperti latihan di kolam renang, relaksasi otot, dan akupunktur.
Perawatan bedah
Tahap penting dari perawatan juga dianggap sebagai metode bedah plastik - operasi yang membantu memperbaiki penampilan pasien. Bedah plastik pada anggota badan dan daun telinga dilakukan, begitu pula pada alat kelamin. Koreksi ginekomastia dengan epispadia juga dilakukan, begitu pula dengan cacat penampilan lainnya.
Pencegahan
Satu-satunya metode pencegahan penyakit ini adalah pemeriksaan pranatal pada ibu hamil. Ada pemeriksaan khusus yang memungkinkan deteksi dini patologi, setelah itu dianjurkan untuk mengakhiri kehamilan. Sebagai alternatif, IVF digunakan, yang dapat membantu anak mewarisi kromosom X yang sehat.
Pencegahan pasien bergantung pada apakah mutasi gen muncul kembali atau diwariskan. Untuk ini, diagnostik genetik molekuler dilakukan. Fakta bahwa tes tersebut tidak mengungkap "kromosom X yang rapuh" pada kerabat mendukung "kesegaran" mutasi tersebut, yang berarti risiko memiliki anak dengan sindrom Martin-Bell sangat kecil. Dalam keluarga yang memiliki orang sakit, tes tersebut akan membantu menghindari kasus yang berulang.
Ramalan cuaca
Prognosis untuk sindrom Martin-Bell menguntungkan untuk seumur hidup, tetapi tidak untuk pemulihan. Harapan hidup bergantung pada tingkat keparahan penyakit dan cacat terkait. Pasien dapat menjalani kehidupan normal. Pada bentuk sindrom Martin-Bell yang parah, pasien berisiko mengalami cacat seumur hidup.
Harapan hidup
Sindrom Martin Bell tidak memiliki dampak negatif yang serius pada kesehatan, sehingga harapan hidup sebagian besar orang yang telah didiagnosis dengan patologi ini tidak berbeda dari indikator standar.