
Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Alkaptonuria adalah kelainan enzim bawaan
Ahli medis artikel

Terakhir ditinjau: 04.07.2025
Salah satu kelainan metabolisme yang sangat langka, alkaptonuria, mengacu pada kelainan bawaan pada metabolisme asam amino tirosin.
Sindrom ini juga dapat disebut defisiensi homogentisat oksidase, homogentisinuria, okronosis herediter, atau penyakit urin hitam.[ 1 ]
Epidemiologi
Menurut statistik, tidak lebih dari sembilan kasus alkaptonuria per 1 juta orang. Dan di sebagian besar negara Eropa, ada satu kasus per 100-250 ribu kelahiran hidup.
Di antara negara-negara Eropa, pengecualiannya adalah Slowakia (terutama wilayah barat laut yang relatif kecil), di mana prevalensi alkaptonuria adalah satu kasus per 19.000 bayi baru lahir. Hal ini kemungkinan besar disebabkan oleh fakta bahwa di antara keluarga-keluarga Roma Slowakia yang tinggal di sana, tingkat perkawinan sedarah (perkawinan antar sepupu) adalah yang tertinggi di Eropa: 10-14%. [ 2 ]
Penyebab alkaptonuria
Penyebab pasti alkaptonuria, sebagai kelainan bawaan katabolisme (pemecahan metabolik) tirosin asam amino α aromatik (homosiklik), telah ditetapkan: jenis kelainan metabolik ini merupakan konsekuensi dari mutasi homozigot atau heterozigot majemuk dari salah satu dari ribuan gen pada kromosom 3, lebih tepatnya, gen HGD pada lokus 3q21-q23 pada lengan panjang kromosom. Gen ini mengkodekan urutan nukleotida enzim hati homogentisat-1,2-dioksigenase [ 3 ] (juga disebut oksidase asam homogentisat atau oksidase homogentisat) - metaloprotein yang mengandung zat besi yang diperlukan untuk salah satu tahap pemecahan tirosin dalam tubuh. [ 4 ], [ 5 ]
Dengan demikian, alkaptonuria merupakan kelainan pada enzim homogentisat-1,2-dioksigenase, atau lebih tepatnya merupakan akibat dari kekurangan atau ketiadaan enzim tersebut secara genetik. [ 6 ]
Karena alkaptonuria merupakan defisiensi enzim bawaan, maka alkaptonuria diwariskan sebagai sifat resesif autosomal. Dengan kata lain, agar alkaptonuria dapat terjadi pada anak, kedua orang tua harus memiliki gen enzim yang dimodifikasi, karena masing-masing orang tua hanya mewariskan satu salinan gen dari dua salinan yang ada kepada anaknya.
Menurut data terkini, terdapat lebih dari dua ratus varian modifikasi gen HGD, dan mutasi salah arti, translokasi, dan penyambungan adalah yang paling sering diamati.
Faktor risiko
Satu-satunya faktor risiko untuk mengembangkan enzimopati kongenital ini adalah adanya riwayat keluarga dan pewarisan dua salinan gen HGD yang dimodifikasi, jika orang tua tidak menunjukkan alkaptonuria (risiko penularan anomali adalah 25%), atau salah satu orang tua memiliki kelainan ini. [ 7 ]
Patogenesis
Tirosin berperan penting dalam sintesis protein, produksi kromoprotein – pigmen kulit melanin, serta hormon tiroid dan neurotransmiter katekolamin.
Mekanisme pengaturan jumlah tirosin dalam sel sangat kompleks, dan tubuh menormalkan kelebihan kandungannya dengan memecahnya. Proses katabolisme tirosin, seperti semua asam amino aromatik, bersifat multitahap dan terjadi dalam beberapa tahap. Setiap tahap pemecahan metabolik tirosin terjadi dengan partisipasi enzim tertentu dan pembentukan senyawa antara.
Jadi, pertama-tama asam amino dipecah menjadi para-hidroksifenilpiruvat, yang diubah menjadi alkapton – 2,5-dihidroksifenilasetat atau asam homogentisat. Kemudian alkapton harus diubah menjadi asam maleasetat, tetapi ini tidak terjadi. [ 8 ]
Dan patogenesis alkaptonuria terdiri dari penghentian reaksi biokimia katabolisme tirosin pada tahap pembentukan asam homogentisat: tidak ada enzim yang diperlukan untuk memecahnya – homogentisat oksidase.
Asam homogenisiat tidak digunakan oleh tubuh dan dapat terakumulasi melalui ekskresi melalui ginjal. Selain itu, asam ini dioksidasi menjadi benzoquinoacetate (asam benzoquinoneacetic), yang, dengan mengikat molekul jaringan dan cairan tubuh, membentuk senyawa biopolimer yang berwarna seperti melanin.
Akumulasi produk antara ini dalam jaringan menyebabkan terganggunya struktur kolagen jaringan tulang rawan, yang mengurangi elastisitasnya – dengan munculnya banyak tanda klinis alkaptonuria dan berkembangnya komplikasi.
Gejala alkaptonuria
Alkaptonuria pada bayi baru lahir dan bayi ditandai dengan warna urin yang menjadi gelap. Ketika terkena udara, urin pada popok, popok bayi, dan pakaian dalam berubah menjadi cokelat tua; hal ini disebabkan oleh akumulasi dan pelepasan asam homogentisat, yang dioksidasi menjadi benzoquinoacetate. [ 9 ]
Jika tidak disertai gejala lain, alkaptonuria pada anak kecil sering kali tidak segera dikenali, karena urin dapat berubah menjadi gelap setelah beberapa jam buang air kecil. Menurut beberapa data, hanya seperlima dari anak di bawah usia 12 bulan yang lahir dengan defisiensi enzim ini yang teridentifikasi dalam situasi klinis. Oleh karena itu, sangat penting bagi orang tua untuk memperhatikan perawatan bayi mereka.
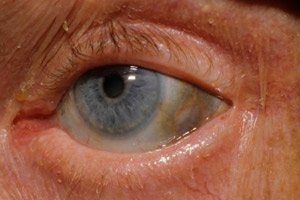
Selain itu, tanda-tanda awal termasuk pigmentasi (warna abu-abu kebiruan) pada sklera mata dan tulang rawan telinga dan hidung, yang sering disebut okronosis.[ 10 ]
Seiring berjalannya waktu, gejala lainnya muncul:
- pigmentasi parah pada kulit di tulang pipi, ketiak dan alat kelamin;
- noda pada pakaian saat bersentuhan dengan area tubuh yang berkeringat;
- serangan kelemahan umum;
- suara serak.
Perlu diingat bahwa alkaptonuria dan okronosis, seperti disebutkan di atas, adalah nama sinonim untuk gangguan katabolisme tirosin yang sama.
Penyakit urin sirup maple dan alkaptonuria. Penyakit urin sirup maple kongenital atau leucinosis juga merupakan kelainan metabolik, memiliki pola pewarisan yang sama, dan bahkan mutasi terjadi pada kromosom yang sama, tetapi memengaruhi gen yang mengkode kompleks enzim dehidrogenase asam α-keto rantai cabang. Karena itu, tubuh tidak dapat memecah komponen protein tertentu, khususnya asam amino leusin, isoleusin, dan valin. Dengan penyakit ini, urin (dan kotoran telinga) memiliki bau yang manis; selain itu, gambaran klinis dari jenis asidosis organik ini meliputi hipopigmentasi, fluktuasi tekanan darah, kejang, muntah dan diare, penurunan kadar glukosa darah, ketoasidosis, halusinasi, dll. Angka kematian pada anak-anak cukup tinggi; pada orang dewasa, tanpa pengobatan, koma dan kematian dapat terjadi karena edema serebral.
Albinisme dan alkaptonuria “disatukan” hanya oleh tirosin. Albinisme, termasuk albinisme okulokutaneus, disebabkan oleh mutasi genetik yang memengaruhi produksi pigmen melanin. Perubahan bawaan terlihat pada gen TYR pada kromosom 11 (11q14.3), yang mengkode tirosinase, enzim melanosom yang mengandung tembaga yang diperlukan untuk pembentukan pigmen kulit berdasarkan produk metabolisme tirosin. Penyakit ini jauh lebih umum daripada alkaptonuria.
Komplikasi dan konsekuensinya
Disebabkan oleh aksi metabolit intermediet tirosin – asam homogentisik dan benzoquinoneasetat – konsekuensi dan komplikasi alkaptonuria muncul karena pengendapan polimer berpigmen reaktif, penghancuran fibril kolagen dan kemunduran kondisi tulang rawan (dengan penurunan ketahanannya terhadap tekanan mekanis).
Selama bertahun-tahun, di masa dewasa, artritis degeneratif dan osteoartritis sendi besar (pinggul, sakroiliaka dan lutut) berkembang; ruang intervertebralis menyempit (terutama di tulang belakang lumbar dan toraks) – dengan kalsifikasi dan pembentukan osteofit; kepadatan jaringan lempeng tulang subkondral menurun, dan tulang-tulang di bawahnya dapat mengalami remodeling patologis dengan pembentukan pertumbuhan dan deformasi. [ 11 ]
Kerusakan pada katup jantung (aorta dan mitral) dan arteri koroner dapat diamati – dengan tanda-tanda penyakit jantung koroner, serta pembentukan batu di ginjal dan kelenjar prostat – karena kalsifikasi yang sama. [ 12 ], [ 13 ]
Diagnostik alkaptonuria
Biasanya, diagnosis kelainan metabolik bawaan didasarkan pada studi cairan biologis tubuh.
Berdasarkan tes dan reaksi apa alkaptonuria dapat didiagnosis? Tes urin diperlukan untuk mendeteksi asam homogentisat dan menentukan kadarnya (normal – 20-30 mg per hari, meningkat – 3-8 g). Sampel urin diperiksa dengan kromatografi gas atau spektrometri massa, menggunakan kromatografi cair; tes skrining untuk keberadaan besi klorida dalam urin dapat dilakukan. [ 14 ]
Ada juga metode untuk diagnostik cepat – menentukan alkapton dalam noda urin kering di atas kertas (berdasarkan intensitas warna).
Dalam memperjelas diagnosis, diagnostik instrumental (radiografi) melibatkan identifikasi tanda-tanda radiologis osteoartritis dan patologi sendi lainnya pada pasien.
Diagnosis dikonfirmasi dengan metode genetik molekuler untuk mendiagnosis penyakit keturunan, seperti pengujian genetik dan pengurutan DNA. [ 15 ]
Perbedaan diagnosa
Diagnosis banding meliputi hemokromatosis dan gagal hati akut pada bayi baru lahir, melaninuria, porfiria intermiten akut, limfohistiositosis hemofagosit, patologi mitokondria primer, artritis reumatoid, ankilosa spondilitis.
Siapa yang harus dihubungi?
Pengobatan alkaptonuria
Pengobatan utama untuk alkaptonuria adalah pemberian asam askorbat dalam dosis besar (minimal 1000 mg per hari) secara oral. Pada anak-anak, hal ini meningkatkan ekskresi asam homogentisat dalam urin, dan pada orang dewasa, hal ini mengurangi kandungan turunannya, asam benzoquinoneacetic, dalam urin, dan memperlambat pengikatannya ke struktur jaringan ikat sendi dan kolagen. [ 16 ]
Klinik-klinik di Eropa Barat sedang menguji obat Nitisinone (Orfalin), obat dari kelompok metabolit yang menghambat tahap kedua katabolisme tirosin: transformasi para-hidroksifenilpiruvat menjadi asam homogentisat. Akan tetapi, penggunaan agen farmakologis ini menyebabkan akumulasi tirosin dan dapat menimbulkan efek samping yang parah, termasuk kekeruhan kornea dan fotofobia, mimisan dan pendarahan lambung, gagal hati, perubahan dalam darah, dll. Meskipun demikian, di Amerika Serikat, Nitisinone telah disetujui oleh FDA untuk pengobatan tirosinemia tipe I. [17 ], [ 18 ]
Oleh karena itu, pengobatan fisioterapi – terapi latihan untuk meningkatkan kekuatan otot dan memperbaiki mobilitas sendi, balneoterapi dan terapi peloid untuk membatasi rasa sakit – dilakukan untuk masalah sendi yang disebabkan oleh alkaptonuria.
Meskipun tirosin tidak hanya diperoleh dari makanan tetapi juga diproduksi di dalam tubuh, penderita alkaptonuria dianjurkan untuk menjalankan diet rendah protein dan membatasi konsumsi makanan yang kaya akan tirosin, terutama daging sapi dan babi, produk susu (terutama keju), kacang-kacangan, kacang-kacangan dan biji-bijian.
Pencegahan
Pencegahan mutasi gen memang tidak mungkin dilakukan, namun untuk mencegah lahirnya anak dengan risiko kelainan bawaan yang tinggi, maka dapat dilakukan konseling genetik medis, yang diperlukan sebelum merencanakan kehamilan bagi pasangan yang memiliki riwayat keluarga dengan penyakit keturunan. [ 19 ]
Ramalan cuaca
Kematian akibat alkaptonuria sangat jarang terjadi, dan kematian dapat disebabkan oleh komplikasi serius yang melibatkan jantung dan ginjal. Jadi, harapan hidup penderita alkaptonuria secara keseluruhan cukup baik.
Namun kualitas hidup berkurang akibat nyeri hebat pada persendian atau tulang belakang dengan keterbatasan mobilitas yang signifikan, seringkali progresif.