
Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Nefritis herediter (sindrom Alport) pada anak-anak
Ahli medis artikel

Terakhir ditinjau: 05.07.2025
Nefritis herediter (sindrom Alport) adalah glomerulopati non-imun herediter yang ditentukan secara genetik, yang dimanifestasikan oleh hematuria (kadang-kadang dengan proteinuria), penurunan fungsi ginjal secara progresif dengan perkembangan gagal ginjal kronis, sering dikombinasikan dengan ketulian sensorineural dan gangguan penglihatan.
Penyakit ini pertama kali dideskripsikan pada tahun 1902 oleh LG Guthrie, yang mengamati sebuah keluarga yang di dalamnya hematuria diamati dalam beberapa generasi. Pada tahun 1915, AF Hurst mendeskripsikan perkembangan uremia pada anggota keluarga yang sama. Pada tahun 1927, A. Alport pertama kali mengidentifikasi gangguan pendengaran pada beberapa kerabat dengan hematuria. Pada tahun 1950-an, lesi mata pada penyakit serupa dideskripsikan. Pada tahun 1972, pada pasien dengan hematuria herediter, selama studi morfologi jaringan ginjal, Hinglais et al. mengungkapkan ekspansi dan stratifikasi yang tidak merata pada membran dasar glomerulus. Pada tahun 1985, dasar genetik nefritis herediter diidentifikasi - mutasi pada gen kolagen tipe IV (Fiengold et al., 1985).
Studi tentang sifat genetik penyakit ini memungkinkan kami untuk menyimpulkan bahwa perbedaan manifestasi fenotipik nefritis herediter (dengan atau tanpa gangguan pendengaran) disebabkan oleh tingkat ekspresi gen mutan. Dengan demikian, saat ini, semua varian klinis dianggap sebagai manifestasi dari satu penyakit dan istilah "nefritis herediter" identik dengan istilah "sindrom Alport".
Menurut penelitian epidemiologi, nefritis herediter terjadi dengan frekuensi 17 per 100.000 anak.
 [
[ Penyebab Sindrom Alport
Dasar genetik penyakit ini adalah mutasi pada gen rantai a-5 kolagen tipe IV. Tipe ini bersifat universal untuk membran basal ginjal, aparatus koklea, kapsul lensa, retina, dan kornea mata, yang telah dibuktikan dalam penelitian menggunakan antibodi monoklonal terhadap fraksi kolagen ini. Baru-baru ini, kemungkinan penggunaan probe DNA untuk diagnostik prenatal nefritis herediter telah ditunjukkan.
Pentingnya pengujian semua anggota keluarga dengan pemeriksaan DNA untuk mengidentifikasi pembawa gen mutan ditekankan, yang sangat penting dalam melakukan konseling medis dan genetik keluarga dengan penyakit ini. Namun, hingga 20% keluarga tidak memiliki kerabat yang menderita penyakit ginjal, yang menunjukkan frekuensi tinggi mutasi spontan gen abnormal. Sebagian besar pasien dengan nefritis herediter memiliki individu dengan penyakit ginjal, gangguan pendengaran, dan patologi penglihatan dalam keluarga mereka; perkawinan sedarah antara orang-orang dengan satu atau lebih nenek moyang penting, karena dalam perkawinan individu yang terkait, kemungkinan menerima gen yang sama dari kedua orang tua meningkat. Rute penularan autosom dominan, autosom resesif, dan dominan, terkait-X telah ditetapkan.
Pada anak-anak, tiga jenis nefritis herediter yang paling sering dibedakan: sindrom Alport, nefritis herediter tanpa gangguan pendengaran, dan hematuria jinak familial.
Sindrom Alport adalah nefritis herediter dengan gangguan pendengaran. Sindrom ini didasarkan pada cacat gabungan dalam struktur kolagen membran dasar glomerulus ginjal, struktur telinga dan mata. Gen sindrom Alport klasik terletak di lokus 21-22 q lengan panjang kromosom X. Dalam kebanyakan kasus, sindrom ini diwariskan secara dominan, terkait dengan kromosom X. Dalam hal ini, sindrom Alport lebih parah pada pria, karena pada wanita fungsi gen mutan dikompensasi oleh alel sehat dari kromosom kedua yang tidak rusak.
Dasar genetik untuk perkembangan nefritis herediter adalah mutasi pada gen rantai alfa kolagen tipe IV. Enam rantai alfa kolagen G tipe IV diketahui: gen rantai a5 dan a6 (Col4A5 dan Col4A5) terletak pada lengan panjang kromosom X di zona 21-22q; gen rantai a3 dan a4 (Col4A3 dan Col4A4) berada pada kromosom ke-2; gen rantai a1 dan a2 (Col4A1 dan Col4A2) berada pada kromosom ke-13.
Dalam sebagian besar kasus (80-85%), pola pewarisan terkait kromosom X dari penyakit ini terdeteksi, yang dikaitkan dengan kerusakan gen Col4A5 sebagai akibat dari delesi, mutasi titik, atau gangguan penyambungan. Saat ini, lebih dari 200 mutasi gen Col4A5 telah ditemukan, yang bertanggung jawab atas terganggunya sintesis rantai a5 kolagen tipe IV. Dengan jenis pewarisan ini, penyakit ini memanifestasikan dirinya pada anak-anak dari kedua jenis kelamin, tetapi pada anak laki-laki lebih parah.
Mutasi pada lokus gen Col4A3 dan Col4A4 yang bertanggung jawab atas sintesis rantai a3 dan a4 kolagen tipe IV diwariskan secara autosom. Menurut penelitian, tipe pewarisan dominan autosom diamati pada 16% kasus nefritis herediter, dan tipe resesif autosom diamati pada 6% pasien. Sekitar 10 varian mutasi gen Col4A3 dan Col4A4 diketahui.
Mutasi ini mengakibatkan terganggunya proses perakitan kolagen tipe IV, yang menyebabkan terganggunya strukturnya. Kolagen tipe IV merupakan salah satu komponen utama membran dasar glomerulus, aparatus koklea, dan lensa mata, yang patologinya akan terdeteksi di klinik nefritis herediter.
Kolagen tipe IV, yang merupakan bagian dari membran dasar glomerulus, terutama terdiri dari dua rantai a1 (IV) dan satu rantai a2 (IV), dan juga mengandung rantai a3, a4, a5. Paling sering, dalam pewarisan terkait kromosom X, mutasi gen Col4A5 disertai dengan tidak adanya rantai a3, a4, a5, dan a6 dalam struktur kolagen tipe IV, dan jumlah rantai o1 dan a2 dalam membran dasar glomerulus meningkat. Mekanisme fenomena ini tidak jelas, diasumsikan bahwa penyebabnya adalah perubahan pasca-transkripsi pada mRNA.
Tidak adanya rantai a3, a4, dan a5 dalam struktur kolagen tipe IV pada membran dasar glomerulus menyebabkan penipisan dan kerapuhannya pada tahap awal sindrom Alport, yang secara klinis lebih sering dimanifestasikan oleh hematuria (lebih jarang oleh hematuria dengan proteinuria atau hanya proteinuria), gangguan pendengaran, dan lentikonus. Perkembangan penyakit lebih lanjut menyebabkan penebalan dan gangguan permeabilitas membran dasar pada tahap akhir penyakit, dengan proliferasi kolagen tipe V dan VI di dalamnya, yang dimanifestasikan dalam peningkatan proteinuria dan penurunan fungsi ginjal.
Sifat mutasi yang mendasari nefritis herediter sangat menentukan manifestasi fenotipiknya. Dalam kasus delesi kromosom X dengan mutasi simultan gen Col4A5 dan Col4A6 yang bertanggung jawab atas sintesis rantai a5 dan a6 kolagen tipe IV, sindrom Alport dikombinasikan dengan leiomiomatosis esofagus dan genital. Menurut data penelitian, dalam kasus mutasi gen Col4A5 yang terkait dengan delesi, tingkat keparahan proses patologis yang lebih besar dicatat, kombinasi kerusakan ginjal dengan manifestasi ekstrarenal dan perkembangan awal gagal ginjal kronis, dibandingkan dengan mutasi titik gen ini.
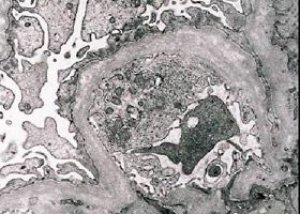
Secara morfologis, mikroskopi elektron menunjukkan penipisan dan stratifikasi membran dasar glomerulus (terutama lamina densa) dan keberadaan granula padat elektron. Lesi glomerulus mungkin heterogen pada pasien yang sama, dari lesi mesangial fokal minimal hingga glomerulosklerosis. Glomerulitis pada sindrom Alport selalu bersifat imunonegatif, yang membedakannya dari glomerulonefritis. Ciri-ciri khasnya meliputi perkembangan atrofi tubulus, infiltrasi limfohistiosit, dan keberadaan "sel busa" dengan inklusi lipid - lipofag. Seiring perkembangan penyakit, penebalan dan kerusakan nyata membran dasar glomerulus terungkap.
Perubahan tertentu dalam sistem imun terungkap. Pasien dengan nefritis herediter memiliki kadar Ig A yang menurun dan kecenderungan untuk meningkatkan konsentrasi IgM dalam darah, kadar IgG dapat meningkat pada tahap awal penyakit dan menurun pada tahap selanjutnya. Mungkin, peningkatan konsentrasi IgM dan G merupakan semacam reaksi kompensasi dalam menanggapi defisiensi IgA.
Aktivitas fungsional sistem limfosit T berkurang; terjadi penurunan selektif limfosit B yang bertanggung jawab terhadap sintesis Ig A, hubungan fagositosis imunitas terganggu, terutama karena terganggunya proses kemotaksis dan pencernaan intraseluler pada neutrofil.
Saat memeriksa biopsi ginjal pada pasien dengan sindrom Alport, data mikroskop elektron mengungkap perubahan ultrastruktur pada membran dasar glomerulus: penipisan, gangguan struktur, dan pemisahan membran dasar glomerulus dengan perubahan ketebalan dan kontur yang tidak rata. Pada tahap awal nefritis herediter, defek tersebut menentukan penipisan dan kerapuhan membran dasar glomerulus.
Penipisan membran glomerulus merupakan tanda yang lebih baik dan lebih umum terjadi pada anak perempuan. Tanda mikroskopis elektron yang lebih konstan pada nefritis herediter adalah terbelahnya membran dasar, dan tingkat keparahan kerusakannya berkorelasi dengan tingkat keparahan prosesnya.
Gejala Sindrom Alport pada Anak
Gejala pertama sindrom Alport berupa sindrom urin terisolasi paling sering terdeteksi pada anak-anak usia tiga tahun pertama. Dalam kebanyakan kasus, penyakit ini terdeteksi secara kebetulan. Sindrom urin terdeteksi selama pemeriksaan pencegahan anak, sebelum masuk ke fasilitas penitipan anak atau selama ARVI. Jika terjadi patologi dalam urin selama ARVI. Pada nefritis herediter, tidak seperti glomerulonefritis yang didapat, tidak ada periode laten.
Pada tahap awal penyakit, kesehatan anak sedikit terganggu, ciri khasnya adalah sindrom urin yang persisten dan resisten. Salah satu tanda utamanya adalah hematuria dengan berbagai tingkat keparahan, yang diamati pada 100% kasus. Peningkatan derajat hematuria dicatat selama atau setelah infeksi pernapasan, aktivitas fisik atau setelah vaksinasi pencegahan. Proteinuria dalam kebanyakan kasus tidak melebihi 1 g / hari, pada awal penyakit bisa tidak konstan, seiring berjalannya proses, proteinuria meningkat. Secara berkala, leukosituria dengan dominasi limfosit dapat hadir dalam sedimen urin, yang dikaitkan dengan perkembangan perubahan interstisial.
Selanjutnya, fungsi ginjal parsial terganggu, kondisi umum pasien memburuk: keracunan, kelemahan otot, hipotensi arteri, sering kali gangguan pendengaran (terutama pada anak laki-laki), dan terkadang gangguan penglihatan muncul. Keracunan dimanifestasikan oleh pucat, kelelahan, dan sakit kepala. Pada tahap awal penyakit, gangguan pendengaran dalam banyak kasus hanya terdeteksi oleh audiografi. Gangguan pendengaran pada sindrom Alport dapat terjadi pada periode yang berbeda di masa kanak-kanak, tetapi paling sering gangguan pendengaran didiagnosis pada usia 6-10 tahun. Gangguan pendengaran pada anak-anak dimulai dengan frekuensi tinggi, mencapai tingkat yang signifikan dalam konduksi udara dan tulang, beralih dari gangguan pendengaran konduksi suara menjadi gangguan pendengaran persepsi suara. Gangguan pendengaran dapat menjadi salah satu gejala pertama penyakit dan dapat mendahului sindrom urin.
Pada 20% kasus, pasien dengan sindrom Alport mengalami perubahan pada organ penglihatan. Kelainan yang paling sering terdeteksi adalah kelainan lensa: sferofokia, lentikonus anterior, posterior atau campuran, dan berbagai katarak. Pada keluarga dengan sindrom Alport, terdapat frekuensi miopia yang signifikan. Sejumlah peneliti terus-menerus mencatat perubahan perimakula bilateral pada keluarga ini dalam bentuk granulasi keputihan atau kekuningan terang pada korpus luteum. Mereka menganggap tanda ini sebagai gejala konstan yang memiliki nilai diagnostik tinggi pada sindrom Alport. KS Chugh dkk. (1993) dalam sebuah penelitian oftalmologi menemukan pada pasien dengan sindrom Alport terjadi penurunan ketajaman penglihatan pada 66,7% kasus, lentikonus anterior pada 37,8%, bintik retina pada 22,2%, katarak pada 20%, dan keratokonus pada 6,7%.
Pada beberapa anak dengan nefritis herediter, terutama bila terjadi gagal ginjal, terlihat adanya keterlambatan perkembangan fisik yang signifikan. Seiring dengan perkembangan gagal ginjal, hipertensi arteri pun berkembang. Pada anak-anak, kondisi ini lebih sering terdeteksi pada masa remaja dan kelompok usia yang lebih tua.
Pasien dengan nefritis herediter ditandai dengan adanya berbagai (lebih dari 5-7) stigma dismorfogenesis jaringan ikat. Di antara stigma jaringan ikat pada pasien, yang paling umum adalah hipertelorisme mata, langit-langit mulut tinggi, anomali gigitan, bentuk daun telinga yang tidak normal, kelengkungan jari kelingking di tangan, dan "celah sandal" di kaki. Nefritis herediter ditandai dengan keseragaman stigma dismorfogenesis dalam satu keluarga, serta frekuensi distribusi yang tinggi di antara kerabat probandus yang melalui garis keturunannya penyakit tersebut ditularkan.
Pada tahap awal penyakit, penurunan fungsi ginjal parsial yang terisolasi terdeteksi: pengangkutan asam amino, elektrolit, fungsi konsentrasi, asidogenesis, perubahan selanjutnya memengaruhi keadaan fungsional bagian proksimal dan distal nefron dan ditandai dengan gangguan parsial gabungan. Penurunan filtrasi glomerulus terjadi kemudian, lebih sering pada masa remaja. Saat nefritis herediter berkembang, anemia berkembang.
Dengan demikian, nefritis herediter ditandai dengan perjalanan penyakit yang bertahap: pertama, tahap laten atau gejala klinis tersembunyi, yang dimanifestasikan oleh perubahan minimal pada sindrom urinarius, kemudian terjadi dekompensasi proses secara bertahap dengan penurunan fungsi ginjal dengan gejala klinis yang nyata (intoksikasi, astenia, keterlambatan perkembangan, anemia). Gejala klinis biasanya muncul tanpa memandang lapisan reaksi inflamasi.
Nefritis herediter dapat bermanifestasi pada periode usia yang berbeda-beda, tergantung pada aksi gen, yang berada dalam keadaan tertekan hingga waktu tertentu.
Klasifikasi
Ada tiga jenis nefritis herediter
- Pilihan I - secara klinis bermanifestasi sebagai nefritis dengan hematuria, gangguan pendengaran, dan kerusakan mata. Perjalanan nefritis bersifat progresif dengan perkembangan gagal ginjal kronis. Jenis pewarisannya dominan, terkait dengan kromosom X. Secara morfologis, pelanggaran struktur membran dasar, penipisan dan pemisahannya terungkap.
- Pilihan II - secara klinis bermanifestasi sebagai nefritis dengan hematuria tanpa gangguan pendengaran. Perjalanan nefritis bersifat progresif dengan perkembangan gagal ginjal kronis. Jenis pewarisannya dominan, terkait dengan kromosom X. Secara morfologis, penipisan membran dasar kapiler glomerulus (terutama laminadensa) terdeteksi.
- Pilihan III - hematuria familial jinak. Perjalanan penyakitnya baik, gagal ginjal kronis tidak terjadi. Jenis pewarisannya adalah autosom dominan atau autosom resesif. Dengan jenis pewarisan autosom resesif, perjalanan penyakit yang lebih parah terjadi pada wanita.
Diagnosis sindrom Alport
Kriteria berikut diusulkan:
- adanya setidaknya dua pasien nefropati dalam setiap keluarga;
- hematuria sebagai gejala utama nefropati pada proband;
- adanya gangguan pendengaran pada setidaknya satu anggota keluarga;
- perkembangan gagal ginjal kronis pada satu atau lebih kerabat.
Dalam diagnosis berbagai penyakit keturunan dan bawaan, tempat yang luas diberikan untuk pendekatan pemeriksaan yang komprehensif dan, terutama, memperhatikan data yang diperoleh saat menyusun silsilah anak. Diagnosis sindrom Alport dianggap valid dalam kasus-kasus di mana 3 dari 4 tanda khas terdeteksi pada pasien: adanya hematuria dan gagal ginjal kronis dalam keluarga, adanya gangguan pendengaran neurosensori, patologi penglihatan pada pasien, deteksi tanda-tanda pembelahan membran dasar glomerulus dengan perubahan ketebalannya dan kontur yang tidak rata selama karakteristik mikroskopis elektron dari biopsi.
Pemeriksaan pasien harus mencakup metode penelitian klinis dan genetik; studi yang ditargetkan tentang riwayat penyakit; pemeriksaan umum pasien dengan mempertimbangkan kriteria yang signifikan secara diagnostik. Pada tahap kompensasi, patologi dapat dideteksi hanya dengan berfokus pada sindrom seperti adanya beban keturunan, hipotensi, banyak stigma disembriogenesis, perubahan sindrom urin. Pada tahap dekompensasi, gejala ekstrarenal dapat muncul, seperti keracunan parah, astenia, perkembangan fisik yang tertunda, anemia, yang bermanifestasi dan mengintensifkan dengan penurunan fungsi ginjal secara bertahap. Pada sebagian besar pasien, dengan penurunan fungsi ginjal, hal berikut diamati: penurunan asido- dan aminogenesis; 50% pasien mencatat penurunan yang signifikan dalam fungsi sekresi ginjal; rentang fluktuasi terbatas dalam kepadatan optik urin; gangguan ritme filtrasi, dan kemudian penurunan filtrasi glomerulus. Tahap gagal ginjal kronis didiagnosis ketika pasien memiliki peningkatan kadar urea dalam serum darah (lebih dari 0,35 g/l) selama 3-6 bulan atau lebih, dan penurunan filtrasi glomerulus hingga 25% dari norma.
Diagnosis banding nefritis herediter harus dilakukan terutama dengan bentuk hematurik glomerulonefritis yang didapat. Glomerulonefritis yang didapat paling sering memiliki onset akut, periode 2-3 minggu setelah infeksi, tanda-tanda ekstrarenal, termasuk hipertensi sejak hari-hari pertama (pada nefritis herediter, sebaliknya, hipotensi), penurunan filtrasi glomerulus pada awal penyakit, tidak ada gangguan fungsi tubulus parsial, sedangkan pada herediter mereka hadir. Glomerulonefritis yang didapat terjadi dengan hematuria dan proteinuria yang lebih jelas, dengan peningkatan LED. Perubahan khas pada membran dasar glomerulus, karakteristik nefritis herediter, memiliki nilai diagnostik.
Diagnosis banding dari nefropati dismetabolik dilakukan dengan gagal ginjal kronis, dalam keluarga secara klinis ditemukan penyakit ginjal heterogen, dan mungkin ada spektrum nefropati dari pielonefritis hingga urolitiasis. Anak-anak sering mengeluhkan nyeri di perut dan secara berkala saat buang air kecil, dalam sedimen urin - oksalat.
Jika diduga adanya nefritis herediter, pasien harus dirujuk ke departemen nefrologi khusus untuk memperjelas diagnosis.
Apa yang perlu diperiksa?
Bagaimana cara memeriksa?
Tes apa yang dibutuhkan?
Siapa yang harus dihubungi?
Pengobatan sindrom Alport
Regimen ini mencakup pembatasan aktivitas fisik berat dan paparan udara segar. Dietnya lengkap, dengan kadar protein, lemak, dan karbohidrat lengkap yang cukup, dengan mempertimbangkan fungsi ginjal. Yang sangat penting adalah deteksi dan pengobatan fokus infeksi kronis. Obat-obatan berikut digunakan: ATP, kokarboksilase, piridoksin (hingga 50 mg/hari), karnitin klorida. Kursus diberikan 2-3 kali setahun. Untuk hematuria, obat herbal diresepkan - jelatang, jus chokeberry, yarrow.
Ada laporan dalam literatur asing dan domestik tentang pengobatan dengan prednisolon dan penggunaan sitostatika. Namun, sulit untuk menilai efeknya.
Pada gagal ginjal kronis, hemodialisis dan transplantasi ginjal digunakan.
Tidak ada metode terapi spesifik (patogenetik yang efektif) untuk nefritis herediter. Semua tindakan pengobatan ditujukan untuk mencegah dan memperlambat penurunan fungsi ginjal.
Pola makan harus seimbang dan berkalori tinggi, dengan mempertimbangkan kondisi fungsional ginjal. Jika tidak ada gangguan fungsional, pola makan anak harus mengandung cukup protein, lemak, dan karbohidrat. Jika ada tanda-tanda disfungsi ginjal, jumlah protein, karbohidrat, kalsium, dan fosfor harus dibatasi, yang akan menunda perkembangan gagal ginjal kronis.
Aktivitas fisik harus dibatasi; anak-anak disarankan untuk menghindari olahraga.
Kontak dengan pasien yang terinfeksi harus dihindari, risiko timbulnya penyakit pernapasan akut harus dikurangi. Sanitasi fokus infeksi kronis diperlukan. Vaksinasi pencegahan tidak dilakukan untuk anak-anak dengan nefritis herediter, vaksinasi hanya mungkin dilakukan untuk indikasi epidemiologis.
Terapi hormonal dan imunosupresif pada nefritis herediter tidak efektif. Ada indikasi beberapa efek positif (pengurangan proteinuria dan perlambatan perkembangan penyakit) dengan penggunaan siklosporin A dan inhibitor ACE jangka panjang selama beberapa tahun.
Dalam pengobatan pasien, obat-obatan digunakan untuk meningkatkan metabolisme:
- piridoksin - 2-3 mg/kg/hari dalam 3 dosis selama 4 minggu;
- kokarboksilase - 50 mg intramuskular setiap hari, total 10-15 suntikan;
- ATP - 1 ml intramuskular dua hari sekali, 10-15 suntikan;
- vitamin A - 1000 IU/tahun/hari dalam 1 dosis selama 2 minggu;
- Vitamin E - 1 mg/kg/hari dalam 1 dosis selama 2 minggu.
Jenis terapi ini membantu memperbaiki kondisi umum pasien, mengurangi disfungsi tubular dan dilakukan secara 3 kali setahun.
Levamisol dapat digunakan sebagai imunomodulator - 2 mg/kg/hari 2-3 kali seminggu dengan jeda antar dosis 3-4 hari.
Menurut data penelitian, oksigenasi hiperbarik memiliki efek positif terhadap tingkat keparahan hematuria dan disfungsi ginjal.
Metode yang paling efektif untuk mengobati nefritis herediter adalah transplantasi ginjal tepat waktu. Dalam kasus ini, tidak ada kekambuhan penyakit pada transplantasi; dalam persentase kecil kasus (sekitar 5%), nefritis dapat berkembang pada ginjal yang ditransplantasi yang terkait dengan antigen pada membran dasar glomerulus.
Arah yang menjanjikan adalah diagnostik prenatal dan terapi rekayasa genetika. Percobaan pada hewan menunjukkan efisiensi tinggi dalam mentransfer gen normal yang bertanggung jawab atas sintesis rantai kolagen alfa tipe IV ke jaringan ginjal, setelah itu sintesis struktur kolagen normal diamati.
Ramalan
Prognosis untuk nefritis herediter selalu serius.
Kriteria prognosis yang tidak menguntungkan untuk perjalanan nefritis herediter adalah:
- jenis kelamin laki-laki;
- perkembangan awal gagal ginjal kronis pada anggota keluarga;
- proteinuria (lebih dari 1 g/hari);
- penebalan membran dasar glomerulus menurut mikroskop;
- neuritis akustik;
- penghapusan pada gen Col4A5.
Prognosis untuk hematuria familial jinak lebih baik.