
Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Lissencephaly pada otak
Ahli medis artikel

Terakhir ditinjau: 04.07.2025
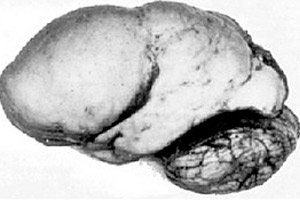
Di antara patologi otak organik, anomali kongenital perkembangan otak seperti lissencephaly menonjol, yang intinya terletak pada permukaan korteks hemisfernya yang hampir halus - dengan jumlah lilitan dan alur yang tidak mencukupi. [ 1 ]
Bila tidak ada konvolusi sama sekali, maka disebut agyria, dan adanya beberapa konvolusi datar yang lebar disebut pachygyria. Cacat ini, seperti beberapa deformasi reduksi otak lainnya, memiliki kode Q04.3 dalam ICD-10.
Epidemiologi
Menurut statistik penyakit langka, terdapat 1-1,2 kasus lissencephaly per 100 ribu bayi baru lahir. [ 2 ], [ 3 ]
Menurut beberapa data, hingga 25-30% kasus lissencephaly klasik diamati pada anak-anak dengan sindrom Miller-Dieker; mutasi titik dan penghapusan gen LIS1 dan DCX terdeteksi pada hampir 85% pasien. [ 4 ]
Studi genetik terhadap 17 gen yang berhubungan dengan lissencephaly menunjukkan bahwa mutasi atau deleksi LIS1 terjadi pada 40% pasien, dan 23% berhubungan dengan mutasi DCX, diikuti oleh TUBA1A (5%) dan DYNC1H1 (3%).[ 5 ]
Penyebab lissencephaly
Semua penyebab yang diketahui dari pembentukan korteks serebral (korteks serebri) hampir atau seluruhnya tanpa lilitan dan alur, yang meningkatkan "area kerja" otak manusia dan memastikan "produktivitas" sistem saraf pusat, dikaitkan dengan gangguan dalam perkembangan perinatalnya. Artinya, lissencephaly berkembang pada janin. [ 6 ]
Kegagalan membentuk lapisan korteks serebral janin pada lissencephaly merupakan akibat dari migrasi abnormal neuron pembentuknya atau penghentian dini proses ini.
Proses ini, yang penting untuk histogenesis serebrokortikal, terjadi dalam beberapa tahap dari minggu ke-7 hingga ke-18 kehamilan. Dan, mengingat peningkatan kepekaannya terhadap mutasi genetik, serta berbagai pengaruh fisik, kimia, dan biologis yang negatif, setiap penyimpangan dari norma dapat menyebabkan lokalisasi neuron yang salah dengan kemungkinan pembentukan lapisan materi abu-abu korteks yang menebal tanpa struktur karakteristik. [ 7 ]
Dalam beberapa kasus, lissencephaly pada anak-anak dikaitkan dengan sindrom Miller-Dieker, Walker-Warburg, atau Norman-Roberts.
Baca juga – Cacat perkembangan otak
Faktor risiko
Selain mutasi pada beberapa gen, faktor risiko kelahiran anak dengan cacat parah tersebut antara lain kekurangan oksigen (hipoksia) pada janin; suplai darah ke otak tidak mencukupi (hipoperfusi); kecelakaan serebrovaskular akut berupa stroke perinatal; patologi plasenta; infeksi virus pada ibu hamil (termasuk TORCH); [ 8 ] masalah metabolisme umum dan fungsi tiroid; merokok, alkohol, zat psikotropika dan narkotika; penggunaan sejumlah obat-obatan; peningkatan tingkat radiasi. [ 9 ]
Patogenesis
Tidak semua kasus lissencephaly memiliki patogenesis yang disebabkan oleh kelainan kromosom dan mutasi gen. Namun, beberapa gen diketahui mengkode protein yang memainkan peran penting dalam pergerakan neuroblas dan neuron yang benar di sepanjang sel glia radial - untuk membentuk korteks serebral. Dan mutasi gen ini menyebabkan patologi ini. [ 10 ]
Khususnya, ini adalah mutasi sporadis (tanpa faktor keturunan) dari gen LIS1 pada kromosom 17, yang mengatur protein motor sitoplasma mikrotubulus dynein, serta gen DCX pada kromosom X, yang mengkode protein doublecortin (lissencephalin-X). [ 11 ]. Dalam kasus pertama, spesialis mendefinisikan lissencephaly klasik (tipe I), dalam kasus kedua – terkait kromosom X. [ 12 ]
Ketika gen FLN1, yang mengkode fosfoprotein filamin 1, dihapus, proses migrasi neuronal yang diarahkan mungkin tidak dimulai sama sekali, sehingga menyebabkan tidak adanya konvolusi (agyria). [ 13 ]
Mutasi telah diidentifikasi pada gen CDK5, yang mengkode enzim kinase – katalisator metabolisme intraseluler, yang mengatur siklus sel pada neuron sistem saraf pusat dan memastikan migrasi normal selama pembentukan struktur otak prenatal.
Perubahan abnormal pada gen RELN pada kromosom 7 yang menyebabkan cacat giral kortikal pada sindrom Norman-Roberts mengakibatkan defisiensi glikoprotein ekstraseluler reelin, yang diperlukan untuk mengatur migrasi dan posisi sel induk saraf selama perkembangan korteks serebri.[ 14 ], [ 15 ], [ 16 ]
Gen ARX mengkodekan protein homeobox non-aristalens, sebuah faktor transkripsi yang memainkan peran penting di otak depan dan jaringan lainnya.[ 17 ] Anak-anak dengan mutasi ARX memiliki gejala lain, seperti hilangnya bagian otak (agenesis korpus kalosum), genitalia abnormal, dan epilepsi parah.[ 18 ],[ 19 ]
Beberapa gen telah dikaitkan dengan lissencephaly. Gen-gen ini termasuk VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A, dan CDK5.[ 20 ]
Cytomegalovirus (CMV) telah dikaitkan dengan perkembangan lissencephaly karena berkurangnya suplai darah ke otak janin. Tingkat keparahan infeksi CMV bergantung pada usia kehamilan. Infeksi dini lebih mungkin menyebabkan lissencephaly karena migrasi neuron terjadi pada awal kehamilan.[ 21 ]
Selain itu, mekanisme terjadinya anomali ini meliputi penghentian migrasi neuron yang tidak lengkap atau lebih lambat dari zona generatif periventrikular ke korteks serebral. Dan dalam kasus seperti itu, lissencephaly atau pachygyria yang tidak lengkap berkembang, di mana beberapa alur dan konvolusi lebar terbentuk (tetapi sebagian besar tidak ada).
Gejala lissencephaly
Tanda-tanda pertama patologi ini (tanpa adanya sindrom yang disebutkan sebelumnya) mungkin tidak muncul segera setelah lahir, tetapi setelah satu setengah hingga dua bulan. Dan yang paling sering diamati adalah gejala klinis lissencephaly berikut:
- hipotonia otot, sering dikombinasikan dengan kelumpuhan spastik;
- kejang dan kejang tonik-klonik umum (dalam bentuk opisthotonus);
- keterbelakangan mental dan pertumbuhan yang parah;
- gangguan fungsi neurologis dan motorik.
Masalah menelan menyebabkan kesulitan dalam memberi makan bayi. [ 22 ]
Gangguan neuromotorik tingkat tinggi sering kali bermanifestasi sebagai tetraplegia – kelumpuhan pada seluruh anggota tubuh. Deformasi pada tangan, jari tangan atau kaki mungkin terjadi.
Pada sindrom Norman-Roberts dengan lissencephaly tipe I, anomali kraniofasial diamati: mikrosefali parah, kemiringan dahi rendah dan pangkal hidung lebar menonjol, mata lebar (hiperterorisme), keterbelakangan rahang (mikrognathia). [ 23 ]
Sindrom Miller-Dieker juga dapat dicirikan oleh ukuran kepala yang luar biasa kecil dengan dahi yang lebar dan tinggi serta hidung yang pendek, lekukan di pelipis (depresi bitemporal), serta telinga yang rendah dan cacat.
Sindrom lissencephaly yang parah ditandai oleh mikrosefali, penurunan ukuran bola mata (mikroftalmia), dikombinasikan dengan displasia retina, hidrosefalus obstruktif, dan korpus kalosum yang tidak ada atau hipoplastik.
Komplikasi dan konsekuensinya
Di antara komplikasi anomali ini, spesialis menyebutkan pelanggaran fungsi menelan (disfagia) dan refluks gastroesofageal; epilepsi refrakter (tidak terkontrol); infeksi saluran pernapasan atas yang sering terjadi; pneumonia (termasuk aspirasi kronis).
Bayi dengan lissencephaly mungkin memiliki masalah jantung organik bawaan berupa defek septum atrium atau cacat jantung kompleks dengan sianosis (tetralogi Fallot). [ 24 ]
Konsekuensi dari retardasi pertumbuhan pascanatal dalam sebagian besar kasus mengakibatkan kematian dalam waktu 24 bulan setelah kelahiran.
Diagnostik lissencephaly
Diagnosis dimulai dengan pemeriksaan fisik anak, studi tentang riwayat medis orang tua, dan riwayat kehamilan dan persalinan.
Selama masa kehamilan, tes DNA janin bebas sel, amniosentesis, atau pengambilan sampel vili korionik mungkin diperlukan. [ 25 ] Untuk informasi lebih lanjut, lihat – Diagnosis Prenatal Penyakit Bawaan
Diagnostik instrumental digunakan untuk memvisualisasikan struktur otak dan menilai fungsinya:
- tomografi terkomputasi otak;
- pencitraan resonansi magnetik (MRI) otak;
- elektroensefalogram (EEG). [ 26 ]
Selama kehamilan, lissencephaly pada USG janin setelah 20-21 minggu dapat dicurigai jika tidak adanya alur parieto-oksipital dan kalkarina serta adanya anomali fisura Sylvian pada otak.
Perbedaan diagnosa
Diagnostik diferensial dengan sindrom cacat otak bawaan lainnya dilakukan.
Terdapat lebih dari 20 jenis lissencephaly, yang sebagian besar terbagi dalam 2 kategori utama: lissencephaly klasik (tipe 1) dan lissencephaly cobblestone (tipe 2). Setiap kategori memiliki manifestasi klinis yang serupa tetapi mutasi genetik yang berbeda.[ 27 ]
Pemeriksaan otak pada lisensefali tipe I menunjukkan korteks serebral dengan empat lapisan, bukan enam lapisan seperti pada pasien normal, sedangkan pada lisensefali tipe 2 korteks serebral tidak teratur dan tampak menggumpal atau nodular akibat perpindahan korteks serebral secara menyeluruh oleh gugusan neuron kortikal yang dipisahkan oleh jaringan gliomesenkim. Pasien juga memiliki kelainan otot dan mata.
- Lissencephaly klasik (tipe 1):
- LIS1: Lissencephaly terisolasi dan sindrom Miller-Dieker (lissencephaly berhubungan dengan dismorfisme wajah). [ 28 ]
- LISX1: mutasi gen DCX. Dibandingkan dengan lissencephaly yang disebabkan oleh mutasi LIS1, DCX memperlihatkan korteks enam lapis, bukan empat lapis.
- Lissencephaly terisolasi tanpa cacat genetik lain yang diketahui
- Lissencephaly batu bulat (tipe 2):
- Sindrom Walker-Warburg
- Sindrom Fukuyama
- Penyakit otot, mata dan otak
- Tipe lain tidak dapat dimasukkan ke dalam salah satu dari dua kelompok di atas:
- LIS2: Sindrom Norman-Roberts, mirip dengan lissencephaly tipe I atau sindrom Miller-Dieker, tetapi tanpa penghapusan kromosom 17.
- LIS3
- LISX2
Mikrolisensefali: Kondisi ini merupakan gabungan dari tidak adanya lipatan kortikal normal dan ukuran kepala yang sangat kecil. Anak-anak dengan lisensefali normal memiliki ukuran kepala normal saat lahir. Anak-anak dengan ukuran kepala kecil saat lahir biasanya didiagnosis dengan mikrolisensefali.
Penting juga untuk membedakan antara lissencephaly dan polymicrogyria, yang merupakan cacat perkembangan otak yang berbeda.
Siapa yang harus dihubungi?
Pengobatan lissencephaly
Lisencephaly adalah kelainan organik yang tidak dapat disembuhkan, sehingga pengobatan yang mungkin dilakukan hanyalah pengobatan suportif dan simptomatis. [ 29 ]
Pertama-tama, ini adalah penggunaan obat antikonvulsan dan antiepilepsi, serta pemasangan selang gastrostomi di lambung (jika anak tidak dapat menelan secara mandiri). Pijat bermanfaat.
Dalam kasus hidrosefalus parah, cairan serebrospinal dikeluarkan.
Pencegahan
Para ahli menyarankan agar calon orang tua mencari konseling genetik, dan agar ibu hamil segera mendaftar ke dokter spesialis kandungan dan ginekolog, serta menjalani semua pemeriksaan terjadwal.
Ramalan cuaca
Bagi anak-anak dengan lissencephaly, prognosisnya tergantung pada tingkatnya, tetapi paling sering perkembangan mental anak tidak melebihi tingkat empat hingga lima bulan. Dan semua anak dengan diagnosis ini menderita gangguan psikomotorik yang parah dan epilepsi yang sulit diobati. [ 30 ]
Menurut NINDS (Institut Nasional Gangguan Neurologis dan Stroke Amerika), harapan hidup maksimal bagi penderita lissencephaly adalah sekitar 10 tahun.