
Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Neuroblastoma pada anak-anak: penyebab, diagnosis, pengobatan
Ahli medis artikel

Terakhir ditinjau: 04.07.2025
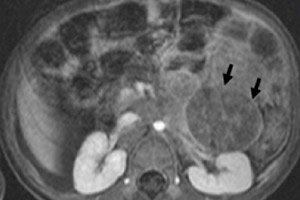
Dalam onkologi pediatrik, salah satu neoplasma ekstrakranial yang paling umum adalah neuroblastoma pada anak-anak, yang merupakan tumor ganas embrionik dari neuroblas krista saraf, yaitu sel saraf embrionik (belum matang) dari sistem saraf simpatik.
Epidemiologi
Menurut statistik dari International Neuroblastoma Risk Group (INRG), neuroblastoma menyumbang sekitar 8% dari semua penyakit onkologis pada anak-anak di seluruh dunia dan menempati urutan ketiga dalam prevalensi, setelah leukemia dan tumor otak.
Menurut data lain, neuroblastoma mencakup sekitar 28% dari semua kanker pada bayi. Lebih dari sepertiga kasus neuroblastoma didiagnosis pada anak di bawah usia satu tahun; usia rata-rata diagnosis adalah 19-22 bulan. Lebih dari 90% kasus yang didiagnosis terjadi pada anak berusia dua hingga lima tahun (dengan dominasi anak laki-laki); insiden puncak diamati pada usia dua hingga tiga tahun, dan kasus pada anak di atas usia lima tahun mencakup kurang dari 10%.
Penyebab neuroblastoma
Dalam mempelajari penyebab neuroblastoma, para peneliti menyimpulkan bahwa tumor pada anak-anak ini terjadi karena mutasi genetik sporadis selama embriogenesis atau perkembangan awal pascanatal. Namun, apa yang menyebabkan perubahan gen ini tidak diketahui, karena tidak ada pengaruh faktor lingkungan teratogenik yang telah diidentifikasi.
Tumor ini dapat terjadi di mana saja, termasuk mediastinum, leher, perut, kelenjar adrenal, ginjal, tulang belakang, dan panggul.
Dalam kasus yang jarang terjadi, neuroblastoma pada bayi dapat dikaitkan dengan mutasi yang diwariskan. Secara khusus, mutasi pada gen protein membran CD246 pada kromosom 2 - enzim tirosin kinase ALK, yang memastikan komunikasi antarsel dan memainkan peran penting dalam fungsi sistem saraf; pada gen protein PHOX2B (pada kromosom 4), yang terlibat dalam pematangan sel saraf.
Neuroblastoma juga dapat dikaitkan dengan neurofibromatosis tipe 1 pada masa kanak-kanak,sindrom Beckwith-Wiedemann, dan hipoglikemia hiperinsulinemia (pankreatitis nesidioblastosis).
Faktor risiko
Saat ini, faktor keturunan diakui sebagai faktor risiko perkembangan neuroblastoma pada anak-anak - adanya tumor ini dalam riwayat keluarga, serta kelainan bawaan yang terkait dengan mutasi gen selama perkembangan intrauterin. Hal ini terutama berlaku untuk kasus perkembangan beberapa neoplasma di berbagai organ.
Tidak ada faktor eksogen yang meningkatkan risiko tumor ini yang telah diidentifikasi oleh para peneliti.
Patogenesis
Mekanisme perkembangan neuroblastoma disebabkan oleh gangguan dalam diferensiasi dan pematangan sel-sel krista neural – garis sel bilateral yang terbentuk di tepi tabung neural dari lapisan germinal ektodermal embrio manusia. Sel-sel ini bermigrasi (bergerak) dan berdiferensiasi menjadi berbagai jenis sel: neuron sensorik dan otonom, sel-sel neuroendokrin dan sel-sel medula adrenal, sel-sel tulang rawan kraniofasial dan tulang, serta sel-sel pigmen.
Pada neuroblastoma, neuroblas yang bermigrasi tidak mengalami pematangan, tetapi terus tumbuh dan membelah, membentuk tumor. Patogenesis pembentukannya dikaitkan dengan mutasi gen berikut:
- dengan duplikasi bagian dari urutan kromosom atau duplikasi segmen gen LMO1 pada kromosom 11, yang mengkode protein RBTN1 dalam sel-sel puncak saraf embrio;
- dengan perubahan jumlah salinan gen NBPF10 pada kromosom 1q21.1, yang mengkode protein DUF1220, yang mengendalikan proliferasi sel induk saraf manusia. Gangguan ini menyebabkan duplikasi kromosom ini atau penghapusannya - tidak adanya bagian DNA;
- dengan perubahan pada gen penekan tumor ATRX (pada kromosom Xq21.1);
- dengan adanya salinan tambahan (amplifikasi) gen faktor transkripsi N-Myc pada kromosom 2, yang mengkode salah satu faktor transkripsi (protein pengikat DNA) yang mengatur aktivitas gen lain dan mengendalikan proliferasi sel prekursor selama pembentukan protein untuk pembentukan jaringan dan organ janin. Amplifikasi gen ini mengubahnya menjadi onkogen, yang memicu gangguan siklus sel, peningkatan proliferasi sel, dan pembentukan tumor.
Gejala neuroblastoma
Tanda-tanda pertama neuroblastoma tidak spesifik dan mungkin meliputi kehilangan nafsu makan (dan penurunan berat badan), kelelahan saat makan, demam, dan nyeri sendi.
Gejala klinis bergantung pada lokasi tumor primer dan keberadaan metastasis (yang terjadi pada 60-73% kasus).
Sangat sering, neuroblastoma primer terlokalisasi di medula adrenal, yang memiliki asal yang sama dengan sel saraf. Pada anak di bawah usia satu tahun, neuroblastoma adrenal didiagnosis pada 35-40% kasus. Gejalanya meliputi nyeri perut, demam, penurunan berat badan, nyeri tulang, anemia atau sindrom Pepper yang menyertainya: kerusakan hati yang menyebar dengan hepatomegali parah dan sindrom gangguan pernapasan.
Neuroblastoma retroperitoneal atau neuroblastoma retroperitoneal pada anak-anak, seiring pertumbuhannya, mulai menekan kandung kemih atau usus, yang dapat menimbulkan masalah buang air kecil atau besar, pembengkakan pada kaki (pada anak laki-laki, skrotum membengkak).
Neuroblastoma mediastinum pada anak-anak (neuroblastoma mediastinum) sering menekan vena cava superior, dan ini dapat menyebabkan pembengkakan pada wajah, leher, lengan, dan dada bagian atas (dengan kulit menjadi merah kebiruan, dengan nodul subkutan). Batuk dan mengi, masalah pernapasan (sesak napas) atau masalah menelan (disfagia) muncul; pembengkakan kelenjar getah bening terlihat di leher, di atas tulang selangka, dan di ketiak.
Penyebaran sel tumor ke sumsum tulang menyebabkan anemia, trombositopenia, dan leukopenia dengan kecenderungan pendarahan.
Dan dengan metastasis di daerah periorbital, lingkaran hitam atau memar muncul di sekitar mata. Tumor semacam itu juga dapat menyebabkan sakit kepala dan pusing, eksoftalmia (bola mata menonjol), dan karena kompresi ujung saraf - kelopak mata terkulai (ptosis) dan penurunan ukuran pupil (miosis).
Neuroblastoma abdomen atau neuroblastoma rongga perut pada anak-anak menyebabkan terbentuknya segel yang dapat diraba di perut, perut membesar, kehilangan nafsu makan, sembelit, dan tekanan darah meningkat. Tumor yang menekan sumsum tulang belakang atau akar saraf dapat menyebabkan mati rasa dan kelemahan anggota badan, ketidakmampuan untuk berdiri, merangkak, atau berjalan. Jika tulang terpengaruh, nyeri tulang dapat terjadi.
Pada kasus tumor stadium 3-4 di rongga perut dengan kerusakan kelenjar getah bening, sel tumor dapat memasuki parenkim ginjal, dan kemudian berkembang menjadi neuroblastoma ginjal yang luas pada anak-anak, yang menyebabkan terganggunya fungsinya.
Tahapan
- Neuroblastoma stadium 1 adalah tumor primer yang terlokalisasi dan terisolasi pada satu area tubuh; kelenjar getah bening di kedua sisi tidak terpengaruh.
- Neuroblastoma stadium 2. Pada stadium 2A, tumor primer terbatas pada satu area tetapi berukuran besar; kelenjar getah bening bilateral tidak terlibat. Pada stadium 2B, kelenjar getah bening di sisi tubuh tempat tumor berada positif mengalami metastasis.
- Neuroblastoma stadium 3: tumor primer melintasi sumsum tulang belakang atau garis tengah tubuh, metastasis unilateral atau bilateral ditemukan di kelenjar getah bening.
- Neuroblastoma stadium 4: tumor telah menyebar ke kelenjar getah bening yang jauh, sumsum tulang, tulang, hati, atau organ lainnya. Dan stadium 4S ditentukan pada anak-anak di bawah usia satu tahun dengan tumor primer yang terlokalisasi, dengan penyebaran ke kulit, hati, atau sumsum tulang.
Sistem Penentuan Stadium Risiko Neuroblastoma Internasional (INRGSS)
INRGSS menggunakan faktor risiko yang ditentukan berdasarkan pencitraan (IDRF), yaitu faktor yang terlihat pada tes pencitraan yang mungkin berarti tumor akan lebih sulit dihilangkan.
INRGSS membagi neuroblastoma menjadi 4 stadium:
- L1: Tumor belum menyebar dari tempat awalnya dan belum tumbuh ke struktur vital. Tumor terbatas pada satu bagian tubuh, seperti leher, dada, atau perut.
- L2: Tumor belum menyebar (bermetastasis) jauh dari tempat awalnya (misalnya, mungkin tumbuh dari sisi kiri perut ke sisi kiri dada), tetapi memiliki setidaknya satu IDRF.
- M: Tumor telah bermetastasis ke bagian tubuh yang jauh (kecuali tumor pada stadium MS).
- MS: Penyakit metastasis pada anak di bawah usia 18 bulan, di mana kanker telah menyebar hanya ke kulit, hati, dan/atau sumsum tulang.
Komplikasi dan konsekuensinya
Neuroblastoma ditandai dengan komplikasi dan konsekuensi seperti:
- penyebaran (metastasis) ke kelenjar getah bening, sumsum tulang, hati, kulit dan tulang;
- kompresi sumsum tulang belakang (yang dapat menimbulkan rasa nyeri dan mengakibatkan kelumpuhan);
- perkembangan sindrom paraneoplastik (disebabkan oleh aksi zat kimia tertentu yang disekresikan oleh tumor, dan juga antigen disialoganglioside GD2 yang diekspresikan oleh sel-selnya), yang dimanifestasikan oleh gerakan mata tak sadar yang cepat, gangguan koordinasi, kram otot, dan diare;
- kambuh setelah selesainya terapi primer (seperti yang ditunjukkan oleh praktik klinis, neuroblastoma berisiko tinggi mengalami kekambuhan pada 50% kasus).
Diagnostik neuroblastoma
Diagnosis dugaan neuroblastoma pada anak memerlukan pemeriksaan, tes laboratorium, dan pencitraan.
Tes darah dan urine dilakukan untuk mengetahui kadar katekolamin (norepinefrin dan dopamin) dan asam homovanilat atau vanililmandelat (terbentuk selama metabolisme hormon-hormon ini); tes darah untuk mengetahui kadar enolase neurospesifik, uji imunosorben terkait enzim (ELISA) serum darah, dan analisis sumsum tulang (sampelnya diambil melalui tusukan aspirasi). Tes DNA dilakukan untuk mengetahui adanya mutasi, dan biopsi dilakukan untuk pemeriksaan sitomorfologi jaringan tumor.
Setelah sampel biopsi diambil, sampel tersebut dikirim ke laboratorium untuk diperiksa di bawah mikroskop oleh seorang ahli patologi (dokter yang memiliki pelatihan khusus dalam mengidentifikasi sel kanker). Tes laboratorium khusus juga sering dilakukan pada sampel untuk menunjukkan apakah tumor tersebut adalah neuroblastoma.
Jika itu neuroblastoma, tes laboratorium juga dapat membantu menentukan seberapa cepat tumor dapat tumbuh atau menyebar, serta perawatan apa yang mungkin paling berhasil.
Diagnostik instrumental memvisualisasikan neoplasma menggunakan USG, sinar X, MRI atau CT, PET dengan pengenalan pemindaian 18F-fluorodeoxyglucose atau MIBG - skintigrafi dengan metaiodobenzylguanidine. [ 1 ]
Perbedaan diagnosa
Diagnosis banding meliputi ganglioneuroma jinak, ganglioneuroblastoma, rhabdomyosarcoma, nefroblastoma.
Siapa yang harus dihubungi?
Pengobatan neuroblastoma
Pada neuroblastoma, pengobatan bergantung pada kelompok risiko pasien (stadium proses tumor), lokasi tumor, fitur genom sel tumor, dan usia anak. Dan pengobatan dapat meliputi pemantauan, pembedahan, kemoterapi, terapi radiasi, imunoterapi, dan transplantasi sel induk hematopoietik.
Kemoterapi neoadjuvan atau adjuvan (pra- atau pascaoperasi) untuk neuroblastoma pada anak-anak, seperti kemoterapi untuk kanker lainnya, diberikan dalam beberapa tahap: obat diberikan selama beberapa hari berturut-turut, diikuti dengan jeda agar tubuh pulih. Siklus biasanya diulang setiap tiga hingga empat minggu.
Obat-obatan berikut (dan kombinasinya) digunakan: Siklofosfamid, Cisplatin atau Karboplatin, Doksorubisin (Adriamisin), Vinkristin, Etoposide.
Efek samping umum dari obat kemoterapi meliputi rambut rontok, kehilangan nafsu makan, kelelahan, mual dan muntah, sariawan, diare, atau sembelit. Kemoterapi dapat merusak sumsum tulang dan menyebabkan penurunan jumlah sel darah.
Imunoterapi tertarget (ditujukan pada antigen tumor GD2) menggunakan obat dari kelompok antibodi monoklonal (anti-GD2 MAb) Dinutuximab (Unituxin) dan Naxitamab. Obat-obatan tersebut diberikan secara intravena melalui infus jangka panjang, dikombinasikan dengan faktor perangsang koloni granulosit-makrofag (sitokin GM-CSF) dan interleukin-2.
Efek samping obat-obatan ini meliputi nyeri (seringkali sangat parah), penurunan tekanan darah, peningkatan denyut jantung, sesak napas (dengan kemungkinan pembengkakan saluran napas), peningkatan suhu, mual, muntah dan diare, perubahan komposisi seluler dan mineral dalam darah.
Untuk mengurangi risiko kekambuhan kanker setelah kemoterapi dosis tinggi dan transplantasi sel induk, anak-anak dengan neuroblastoma berisiko tinggi diobati dengan retinoid sistemik, asam retinoat 13-cis (Isotretinoin). [ 2 ]
Perawatan bedah neuroblastoma – pengangkatan tumor, misalnya adrenalektomi terbuka atau reseksi laparoskopi neuroblastoma adrenal; limfektomi (pengangkatan kelenjar getah bening yang terkena), dll. [ 3 ]
Untuk neuroblastoma berisiko tinggi, terapi radiasi dapat digunakan.[ 4 ]
Pencegahan
Mengingat penyebab neuroblastoma pada anak-anak, satu-satunya tindakan pencegahan mungkin adalah konseling genetik saat merencanakan kehamilan. Namun perlu diingat bahwa tumor ini dikaitkan dengan mutasi bawaan hanya pada 1-2% kasus.
Ramalan cuaca
Neuroblastoma infantil memiliki kemampuan untuk mengalami regresi spontan.
Penanda prognostik
- Tumor berisiko tinggi, serta neuroblastoma pada anak-anak dari semua kelompok usia dan semua stadium (kecuali stadium 4S) – dengan peningkatan ekspresi gen N-MYC dan amplifikasi onkogen N-Myc – memiliki prognosis yang tidak baik sehingga memengaruhi harapan hidup.
- Memiliki sel tumor yang kehilangan bagian tertentu dari kromosom 1 atau 11 (dikenal sebagai delesi 1p atau 11q) memiliki prognosis yang lebih buruk. Memiliki bagian tambahan dari kromosom 17 (peningkatan 17q) juga dikaitkan dengan prognosis yang lebih buruk.
- Sel neuroblastoma dengan jumlah DNA yang besar memiliki prognosis yang lebih baik, terutama untuk anak-anak di bawah usia 2 tahun.
- Neuroblastoma yang memiliki lebih banyak reseptor neurotrofin, terutama reseptor faktor pertumbuhan saraf TrkA, memiliki prognosis yang lebih baik.
Kelangsungan hidup menurut kelompok risiko Childhood Oncology Group (COG)
- Kelompok risiko rendah: Anak-anak dalam kelompok risiko rendah memiliki tingkat kelangsungan hidup 5 tahun lebih besar dari 95%.
- Kelompok risiko menengah: Anak-anak dalam kelompok risiko menengah memiliki tingkat kelangsungan hidup 5 tahun sebesar 90% hingga 95%.
- Kelompok berisiko tinggi: Anak-anak dalam kelompok berisiko tinggi memiliki tingkat kelangsungan hidup 5 tahun sekitar 50%.
Sekitar 15% kematian akibat kanker pada anak disebabkan oleh neuroblastoma. Peluang bertahan hidup jangka panjang untuk keganasan berisiko tinggi ini tidak lebih dari 40%. Tingkat kelangsungan hidup lima tahun secara keseluruhan adalah 67-74%, 43% pada kelompok usia satu hingga empat tahun, dan lebih dari 80% untuk neuroblastoma yang didiagnosis pada tahun pertama kehidupan.